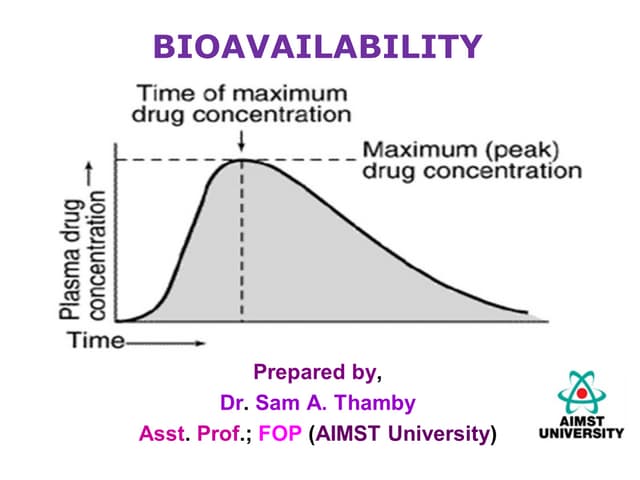
![ BIOAVAILABILITY: means rate & extent of absorption
of unchanged drug from its dosage form or site of
administration to the systemic circulation.
Order: Parentral(i,.v)>oral>rectal>topical
Absolute Bioavailability:
It is the systemic availability of drug after
extravascular administration compared to i.v dosing of
the same drug.
F= [AUC]oral / [AUC]i.v*doseiv/doseoral](https://image.slidesharecdn.com/bioavailibility-112070804016-130119105853-phpapp02/85/Bioavailibility-112070804016-2-320.jpg)
![ Relative Bioavailability:
It is the systemic availability of the drug after
oral administration is compared with that of an
oral standard of the same drug.
Fr=[AUC]test/ [AUC]std*dosetest/dosestd,
where,Fr=Relative Bioavailability](https://image.slidesharecdn.com/bioavailibility-112070804016-130119105853-phpapp02/85/Bioavailibility-112070804016-3-320.jpg)




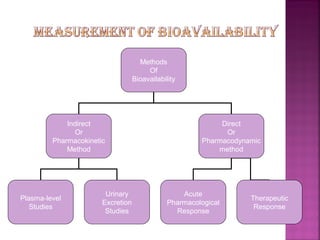
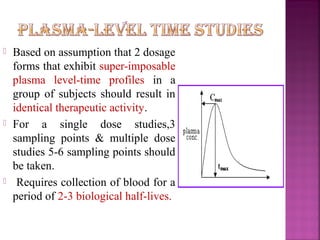


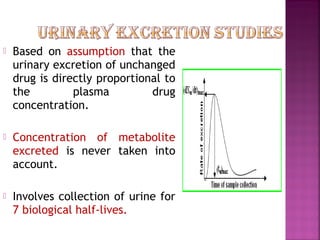


















This document discusses bioavailability and factors affecting it. It defines bioavailability as the rate and extent of absorption of a drug into systemic circulation from its dosage form. Absolute bioavailability is the systemic availability of a drug after extravascular administration compared to intravenous dosing. Relative bioavailability compares the systemic availability of a drug after oral administration to that of an oral standard. The document then discusses primary stages of drug development, factors affecting subject selection in bioavailability studies, and methods of assessing bioavailability including pharmacokinetic, urinary excretion, and pharmacological response methods.