Downloaded 1,040 times

























![ The kinetics of reversible drug–protein binding for a protein with one
simple binding site can be described by the law of mass action, as
follows:
or ………………1
The law of mass action, an association constant, K a, can be
expressed as the ratio of the molar concentration of the products
and the molar concentration of the reactants. This equation
assumes only one-binding site per protein molecule
……………………….…2
Experimentally, both the free drug [D] and the protein-bound drug
[PD], as well as the total protein concentration [P] + [PD], may be
determined. To study the binding behavior of drugs, a determinable
ratio (r )is defined, as follows](https://image.slidesharecdn.com/drugdistributionclearance-140930003146-phpapp02/85/Drug-distribution-clearance-33-320.jpg)
![moles of drug bound is [PD] and the total moles of protein is [P] + [PD], this
equation becomes
Substituting the value of PD from equa.
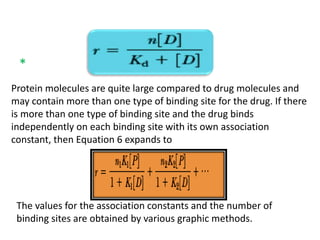
This equation describes the simplest situation, in which 1 mole of drug binds to 1 mole of
protein in a 1:1 complex. This case assumes only one independent binding site for each
molecule of drug. If there are n identical independent binding sites per protein molecule, then
the following is used:](https://image.slidesharecdn.com/drugdistributionclearance-140930003146-phpapp02/85/Drug-distribution-clearance-34-320.jpg)
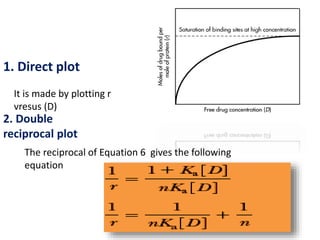
![ A graph of 1/r versus 1/[D] is
called a double reciprocal plot.
The y intercept is 1/n and the
slope is 1/nKa . From this graph ,
the number of binding sites may
be determined from the y
intercept, and the association
constant may be determined from
the slope, if the value for n is
known.
3. Scatchard plot
The Scatchard plot spreads the data to give a
better line for the estimation of the binding
constants and binding sites
r = n Kas – r Kas
[DF]](https://image.slidesharecdn.com/drugdistributionclearance-140930003146-phpapp02/85/Drug-distribution-clearance-37-320.jpg)



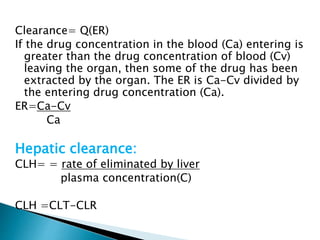








![Model-Independent Methods
*Clearance rates may also be estimated by a single
(nongraphical) calculation from knowledge of the (AUG) 0 to
infinite, the total amount of drug absorbed,FD0, and the
total amount of drug excreted in the urine, D u to infinite.
For example, if a single IV bolus drug injection is given to a
patient and the (AUG) 0 to infinite is obtained from the
plasma drug level-time curve, then total body clearance is
estimated by
Clt= Do
[AUG] 0 to infinite
*If the total amount of drug excreted in the urine D u to
infinite, has been obtained, then renal clearance is
calculated by
Clt= D 0 to infinite
[AUG] 0 to infinite](https://image.slidesharecdn.com/drugdistributionclearance-140930003146-phpapp02/85/Drug-distribution-clearance-52-320.jpg)











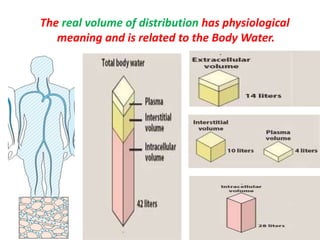
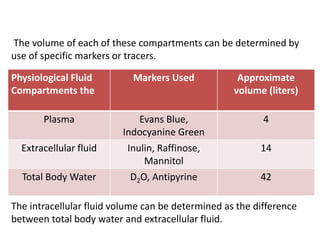
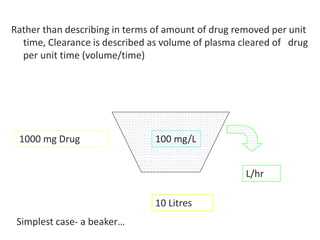
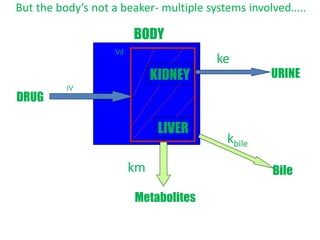
This document provides an overview of drug distribution and clearance. It discusses several key points: 1) Drug distribution refers to the reversible transfer of drugs between compartments in the body, reaching equilibrium between blood and tissues. The rate and extent of distribution provides information about a drug's pharmacokinetics. 2) The volume of distribution is used to quantify how a drug is distributed between plasma and tissues after dosing. It represents the apparent volume required for the total amount of drug administered. 3) Many factors influence a drug's distribution, including physicochemical properties, protein and tissue binding, blood flow rates, and physiological barriers. Highly perfused tissues equilibrate quickly with lipid-soluble drugs.














![Clinical Pharmacokinetics-I [half life, order of kinetics, steady state]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalpk-ihalflifeorderofkineticssteadystate-140217020044-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)




















































