Download as PPSX, PPTX




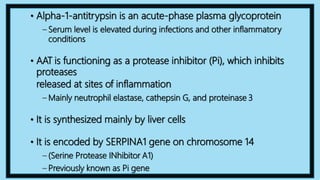


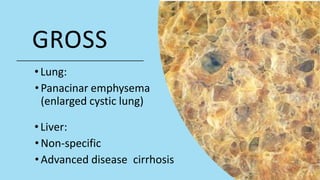
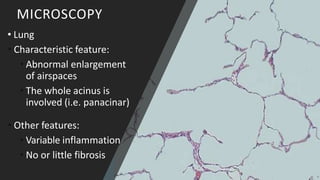
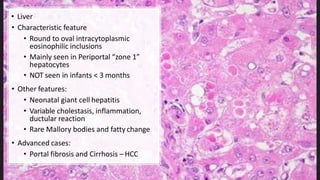
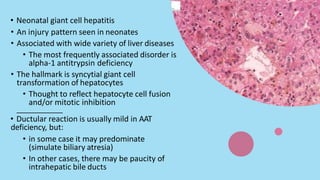

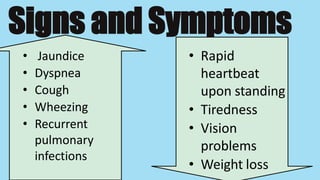


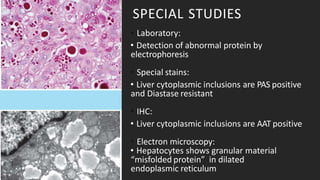

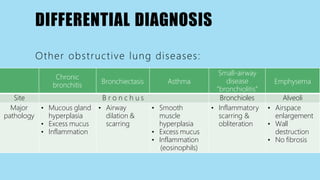
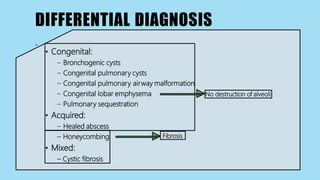

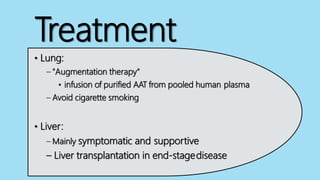


Alpha-1 Antitrypsin Deficiency is an autosomal recessive disease caused by mutations in the SERPINA1 gene resulting in low levels of alpha-1 antitrypsin (AAT) protein. This puts individuals at risk for lung and liver disease. In the lungs, a lack of AAT allows proteases to destroy tissue and cause emphysema. In the liver, mutated AAT accumulates in hepatocytes and can lead to cirrhosis. The disease is diagnosed through blood tests measuring AAT levels and treated through AAT augmentation therapy or liver transplantation for end-stage liver disease.

























![Interstitial Lung Diseases [ILD] Approach to Management](https://cdn.slidesharecdn.com/ss_thumbnails/interstitiallungdiseases-arunvasireddy-19october2015-seminar-171016041856-thumbnail.jpg?width=640&height=640&fit=bounds)







































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)















