Downloaded 102 times




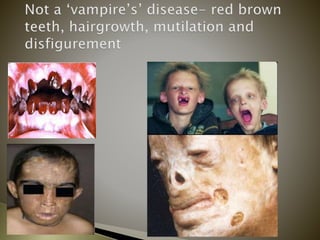

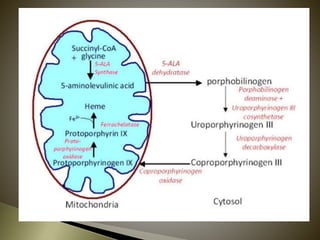
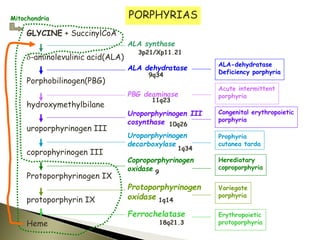

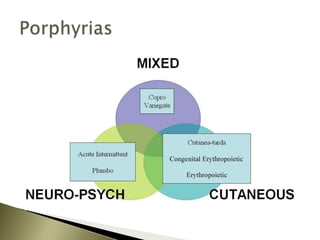









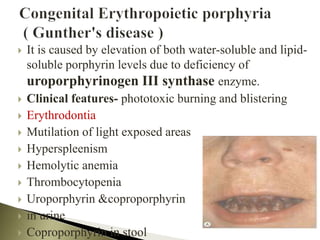


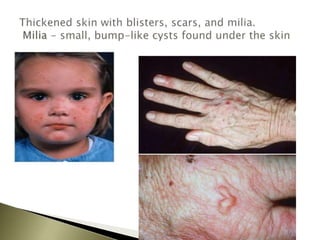
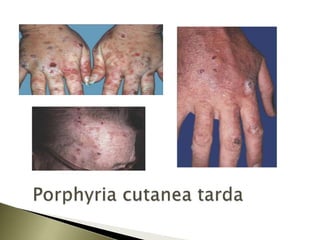







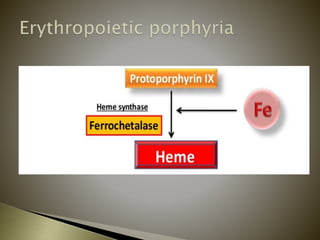

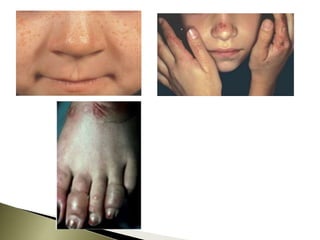

















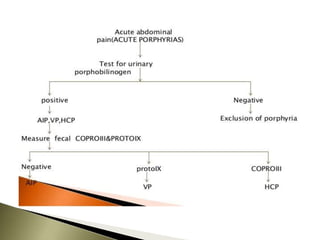



Porphyrias are a group of rare hereditary disorders caused by enzyme deficiencies in the heme biosynthetic pathway, leading to toxic accumulation of heme precursors and various symptoms including photosensitivity, abdominal pain, and neurological issues. Treatments involve management of symptoms, administration of hemin, and avoidance of triggers such as certain medications and foods. Though symptoms can be severe and the condition often misunderstood, porphyrias do not lead to a craving for blood, contrary to common myths.























































































