Downloaded 318 times

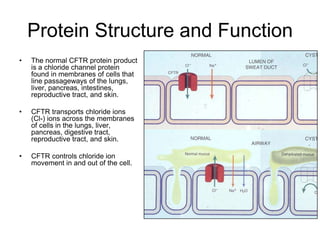










Cystic fibrosis is a genetic disease caused by mutations in the CFTR gene that result in a defective chloride channel protein. This leads to thick, sticky mucus in the lungs and other organs. In the lungs, the mucus clogs the airways and traps bacteria, causing chronic lung infections. While treatments can help manage symptoms, the only cure would be gene therapy to replace the defective gene or provide the normal protein before damage occurs. The goal of treatment is to clear the lungs of mucus and control infections.































































































