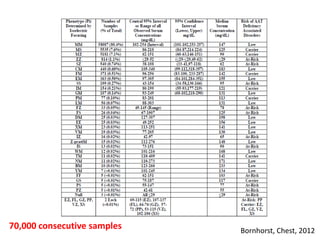
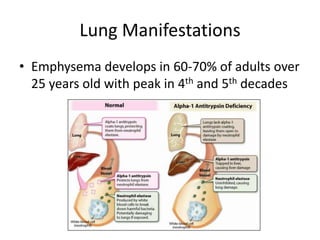
This document discusses alpha 1-antitrypsin (A1AT) deficiency, which can cause liver disease. It presents a case study of an 8 month old boy diagnosed with A1AT deficiency based on his brother's history. The pathophysiology of A1AT deficiency involves accumulation of abnormal protein in liver cells, which can lead to inflammation and fibrosis over many years. While treatment options are limited, monitoring and potential liver transplantation are discussed. The document reviews genetics, symptoms, diagnosis, and novel areas of research on this under-recognized genetic condition.