Downloaded 372 times



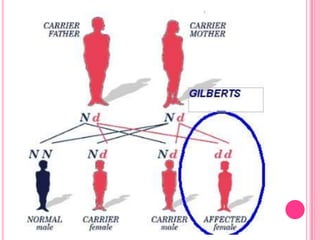









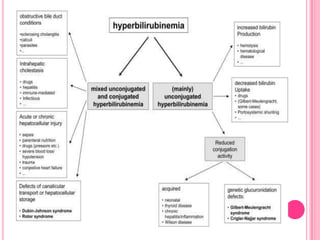








Gilbert syndrome is a mild liver disorder caused by a genetic mutation that reduces the liver's ability to break down bilirubin. This leads to mildly elevated bilirubin levels in the blood and sometimes jaundice. It is inherited in an autosomal dominant pattern and is generally harmless, with no associated morbidity or mortality. The condition is diagnosed based on increased bilirubin levels and can be differentiated from other liver conditions through additional blood tests. Treatment is typically not needed as Gilbert syndrome itself causes no harm.











































































































