Downloaded 52 times






















![Background
Nitrogen (N) is a key macronutrient for plants and has a strong
influence on crop development and productivity.
To increase crop yield, the application of synthetic N fertilizers to
crops has increased substantially in recent decades. However, plants
utilize less than half of the applied N because of low N use efficiency
(NUE) and uptake saturation.
Transgenic plants with improved NUE have been developed in which
expression of protein-coding genes involved in N uptake,
assimilation, and transport have been modulated by genetic
engineering.
Overexpression of nitrate transporters [3–6] or ammonium
transporters [7–10] led to enhanced N source uptake ability and
increased nitrate and ammonium contents in transgenic plants.
Overexpression of N assimilation enzymes, including alanine
aminotransferase (AlaAT) [11, 12] and glutamine synthetase (GS)
increased total N content and plant dry biomass and produced yield
increases.](https://image.slidesharecdn.com/creditseminarsajadahsheikh-220129132428/85/Transcriptomics-A-time-efficient-tool-for-crop-improvement-31-320.jpg)















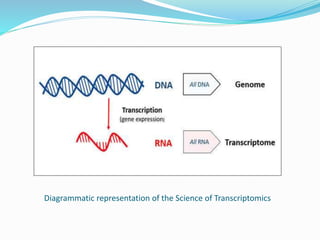
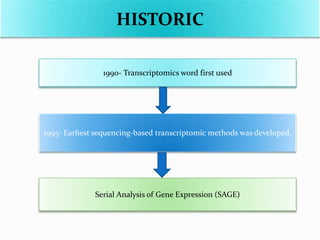
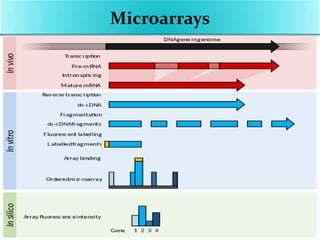
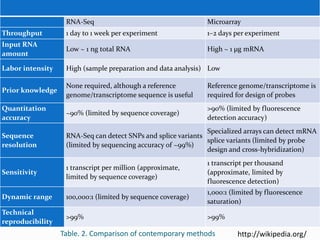
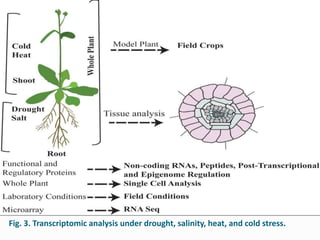
Transcriptomics is the study of an organism's transcriptome, the complete set of RNA transcripts in a cell or tissue under a specific set of conditions. It can be used to identify genes and pathways involved in plant responses to biotic and abiotic stresses like drought, salinity, pathogens, and nutrient deficiencies. Microarrays and RNA-seq are two main techniques used in transcriptomics. Applications of transcriptomics in crop biotechnology include understanding stress responses as well as plant responses to insects and abiotic stresses like salinity and cold which can help develop stress-tolerant crops.


































































