Downloaded 118 times









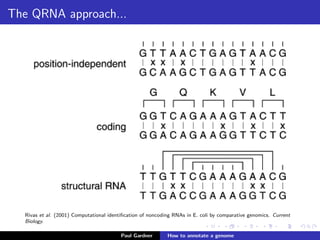


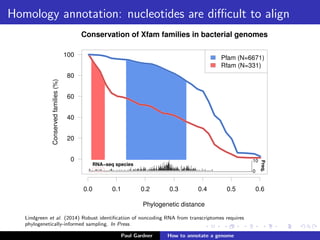




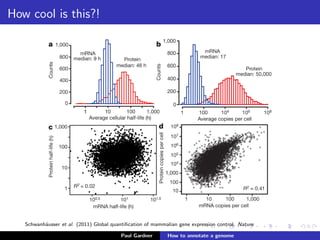
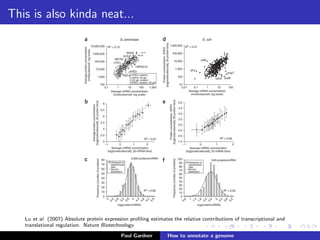





This document discusses various methods for annotating genomes after sequencing and assembly. Sequence analysis approaches like identifying open reading frames can rapidly and inexpensively find some genes, but have weaknesses like false positives and missing short genes. More accurate methods are needed to find non-coding RNAs, pseudogenes, and other elements. As sequencing technologies generate more data, the bottleneck has shifted to analysis, requiring skills in both biology and mathematics. The document provides an example sequence to annotate and poses questions about fast, cheap and accurate annotation methods.











































































































