Downloaded 58 times


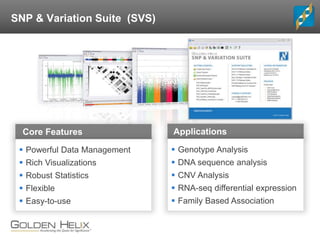


















The document discusses rare variant analysis workflows for next-generation sequencing (NGS) data in large cohorts, highlighting tools and techniques for analyzing rare variants. It covers topics such as the importance of combining rare variants for analysis, various methods for approaching rare variant tests, and the functionalities of the software suite by Golden Helix, including GenomeBrowse and SNP & Variation Suite (SVS). The document emphasizes the need for consistency in NGS analysis and offers practical insights into managing and visualizing genetic data.











































































