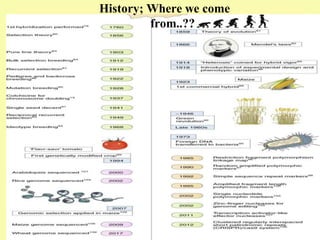
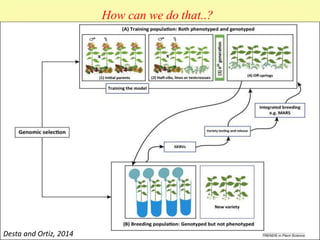
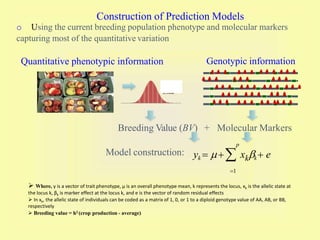
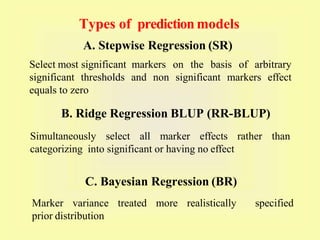
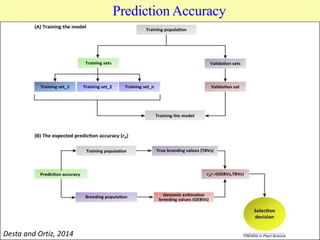
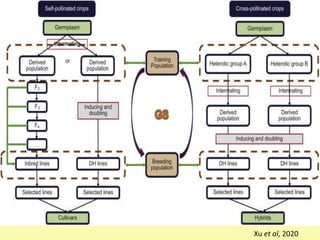
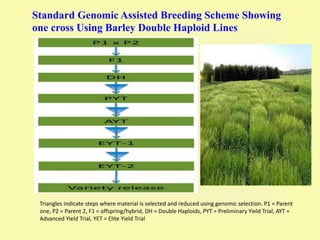
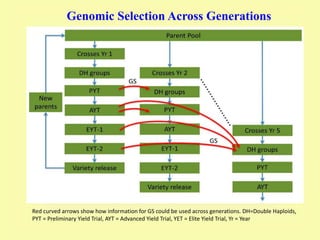
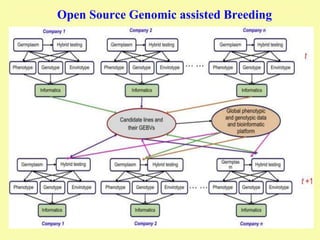
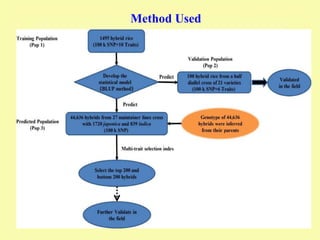
This document discusses genomic selection in plants. It begins with an introduction to genomic selection and its history. Genomic selection uses dense genetic markers and phenotypic data from a reference population to develop prediction equations that can then be applied to other populations to estimate genomic breeding values without additional phenotyping. The document outlines the steps involved, including preparing phenotypic and genotypic data, constructing prediction models, fitting and evaluating models, and applying genomic selection in breeding programs. It provides examples of software used and factors that affect prediction accuracy. The document concludes with two case studies, one on genomic selection for hybrid rice and another on genomic selection to improve wheat grain quality.