Downloaded 163 times

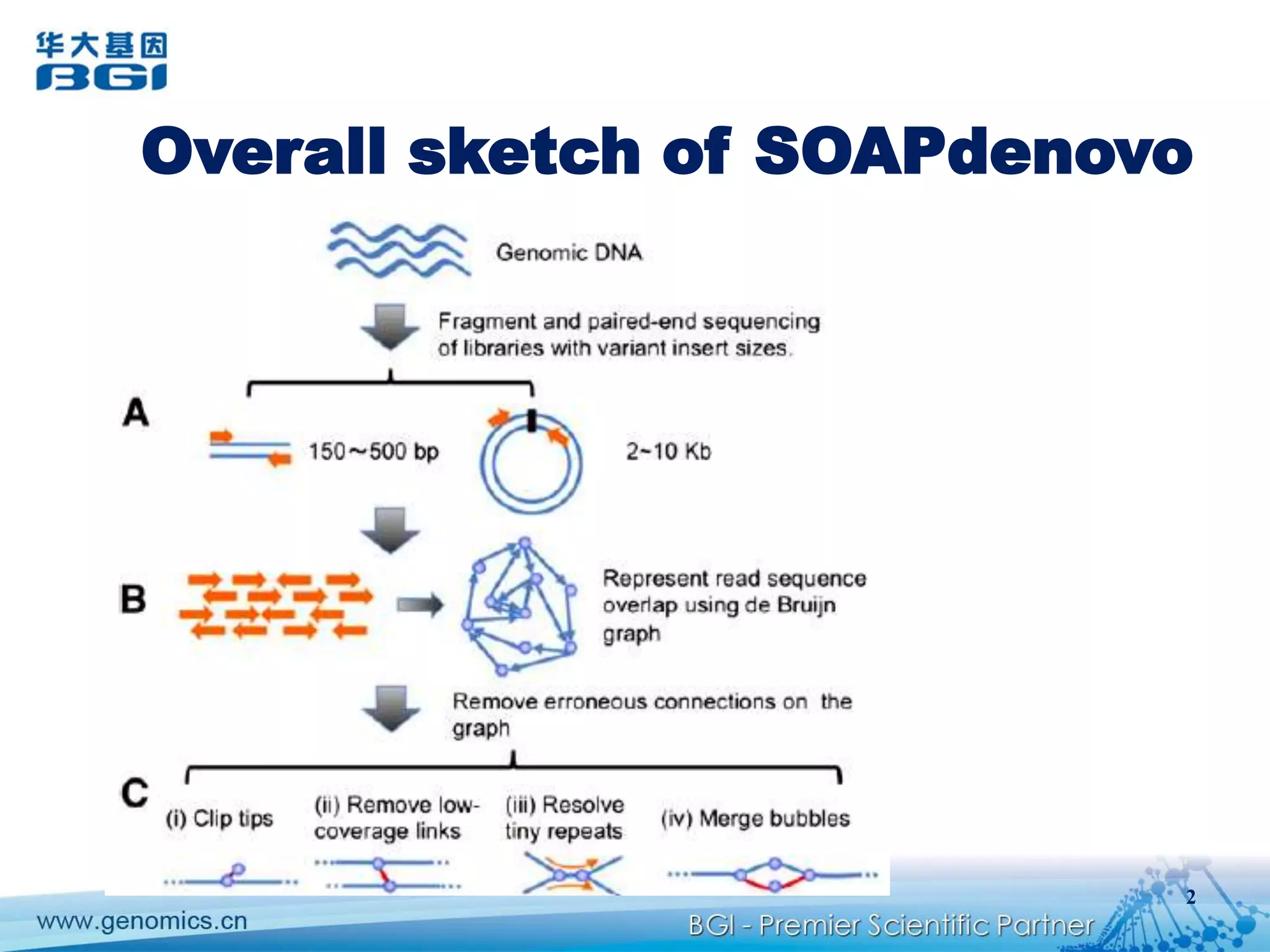
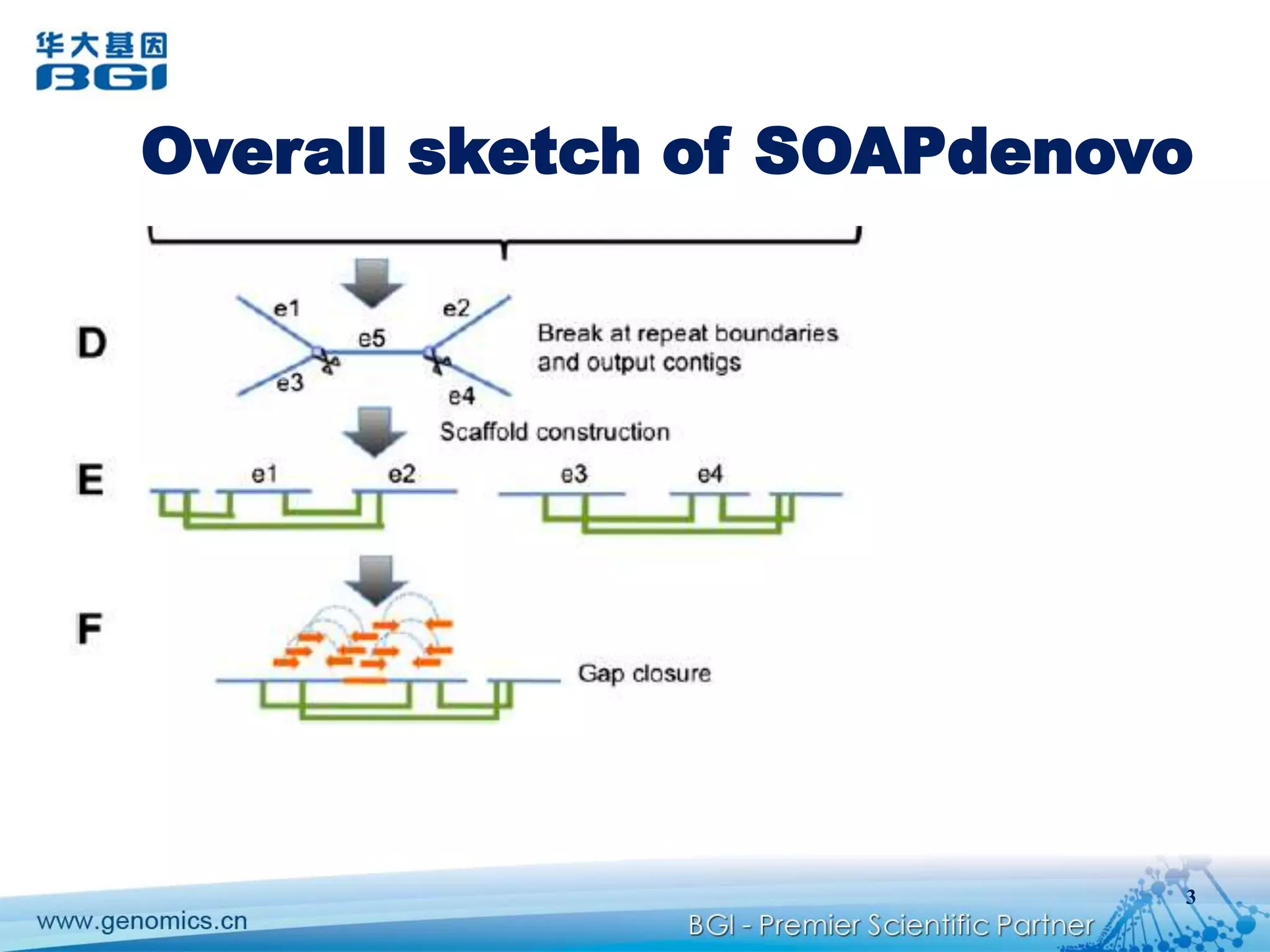



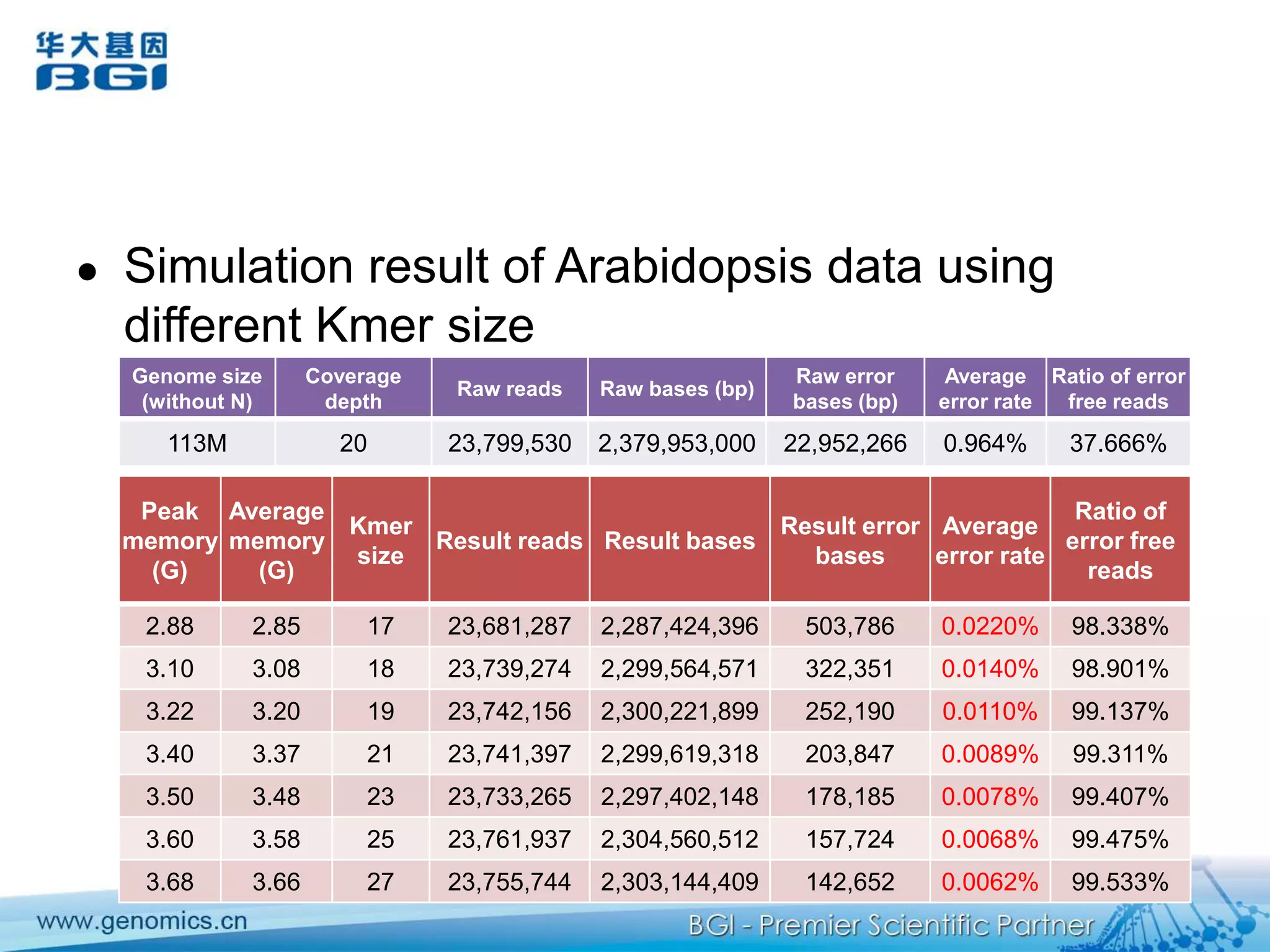







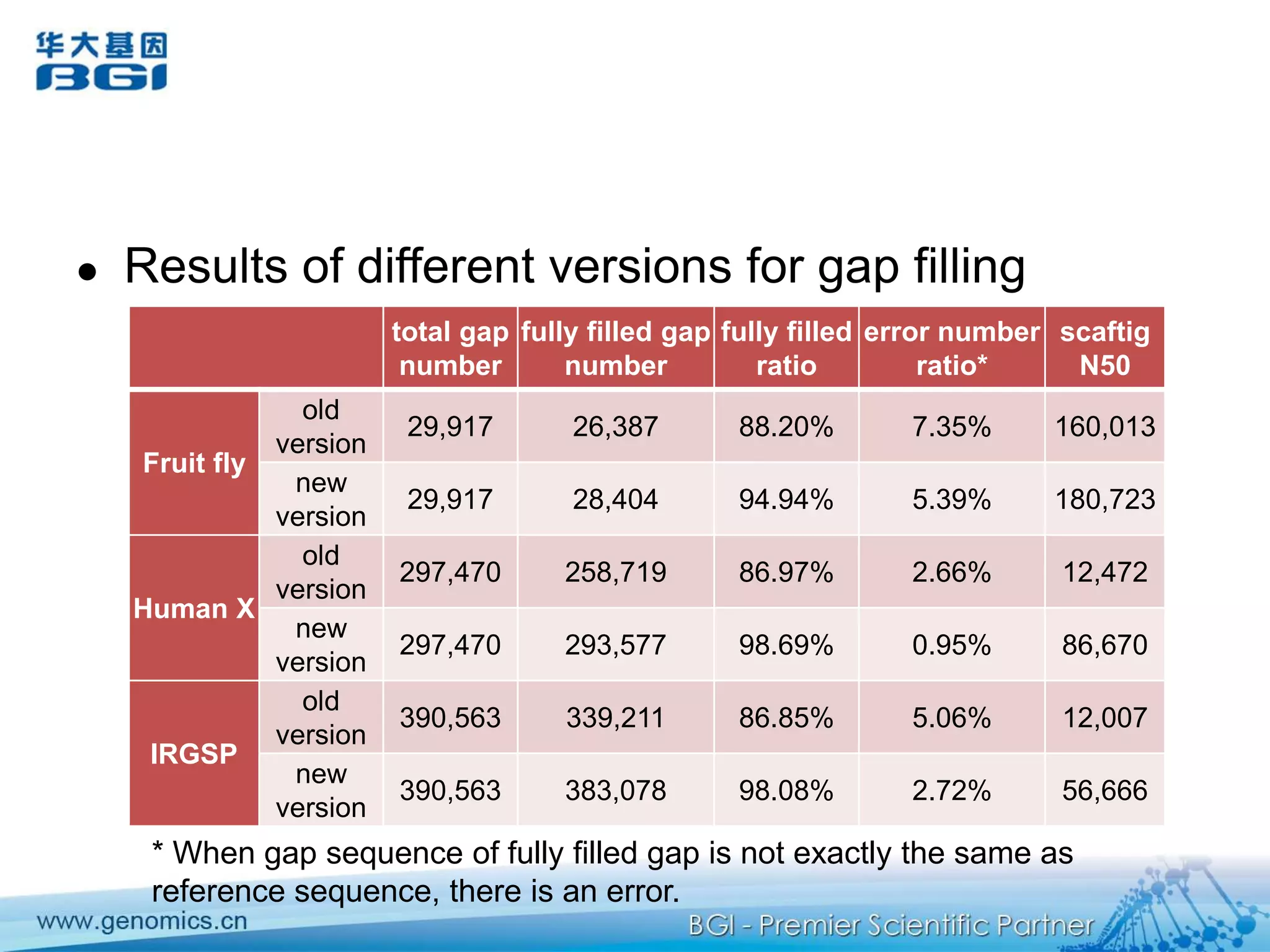




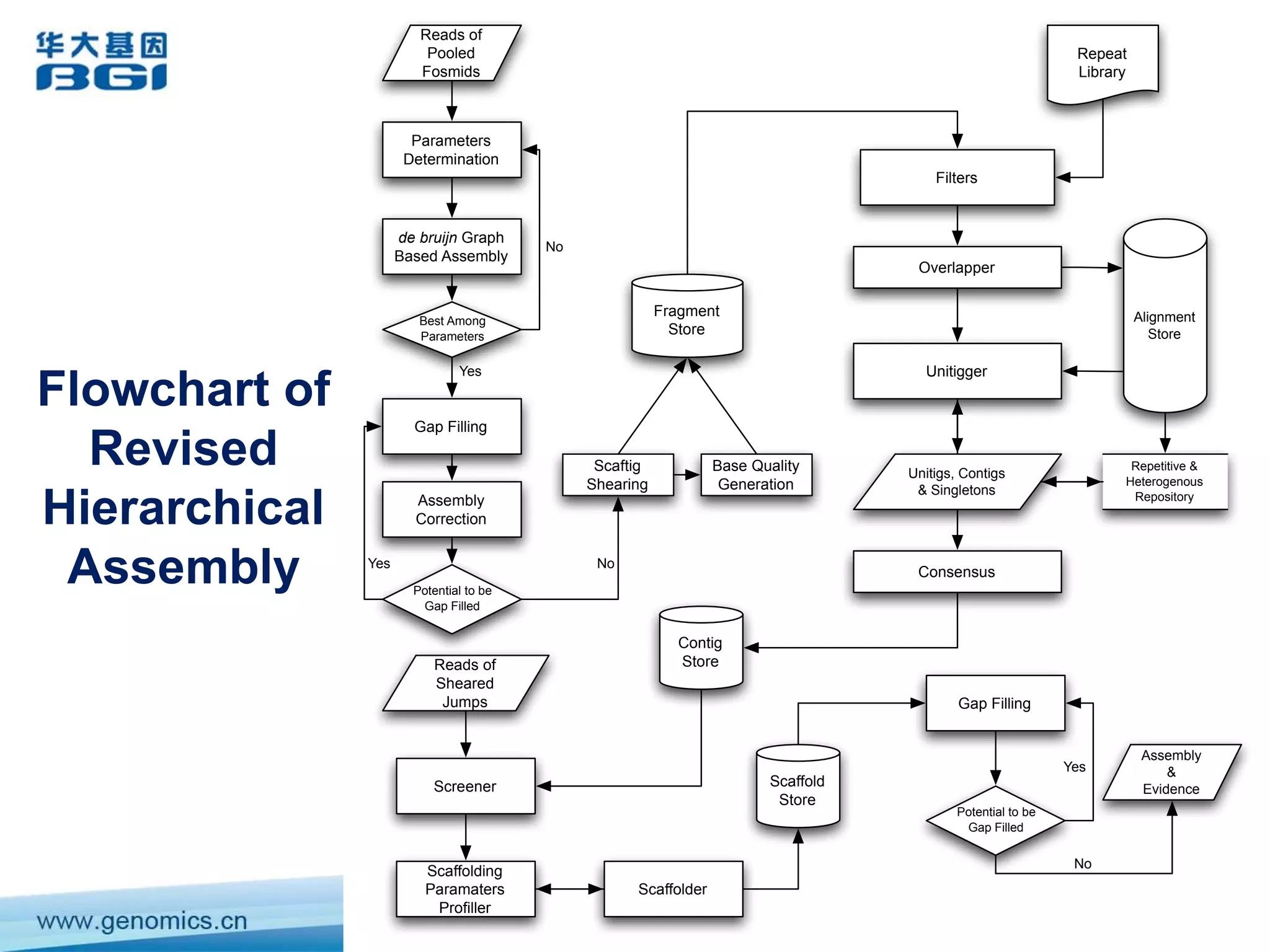



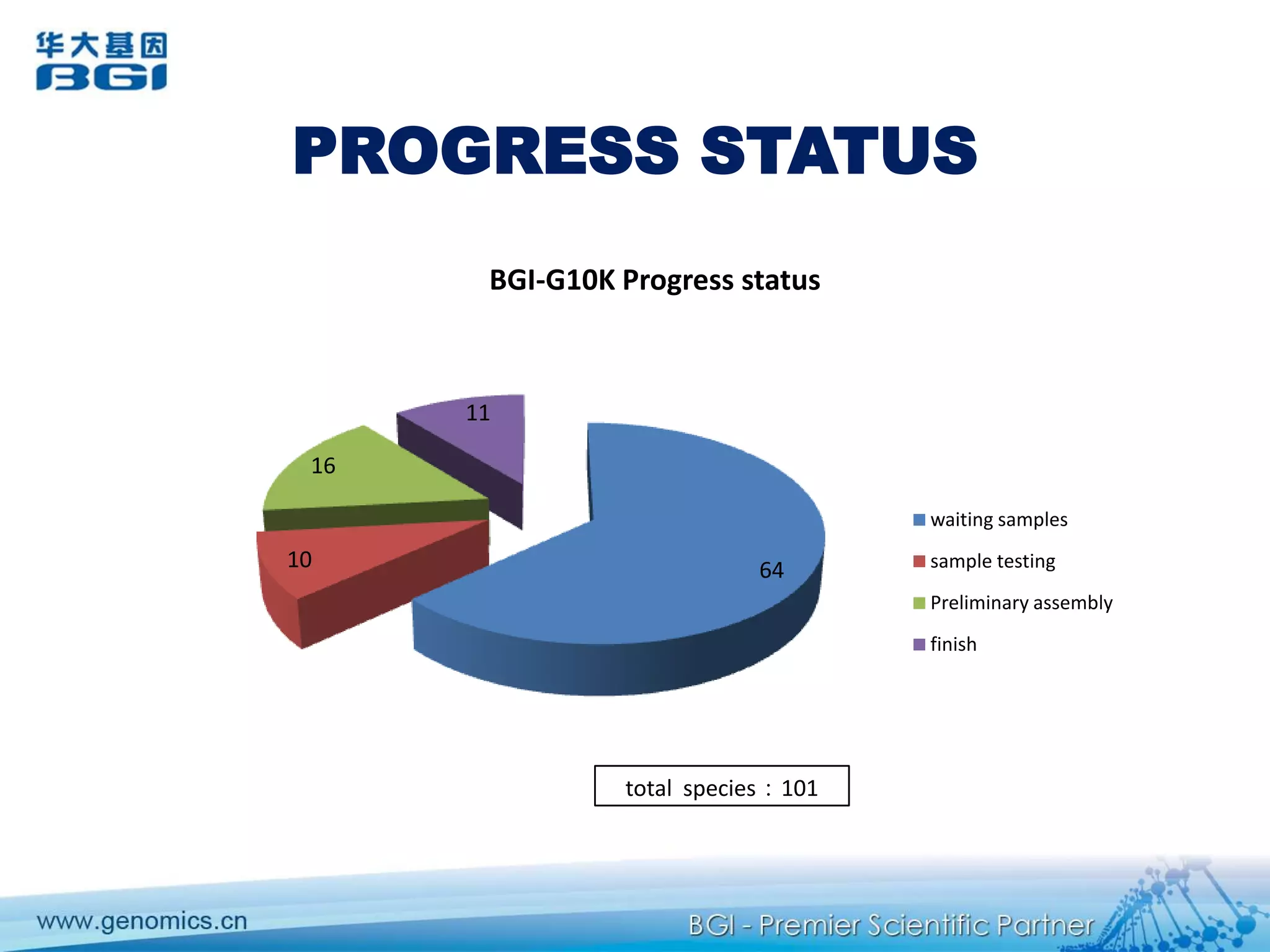
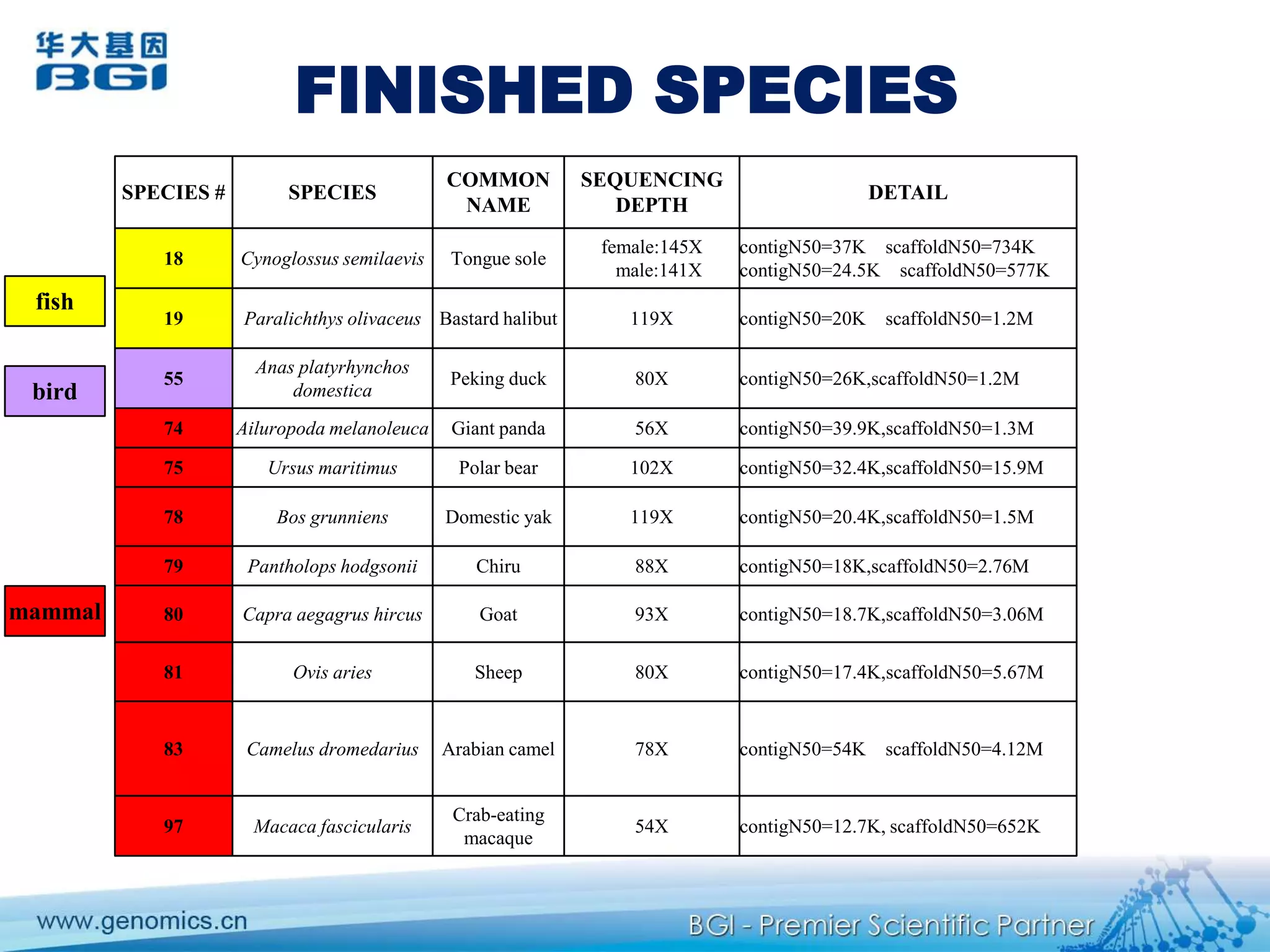
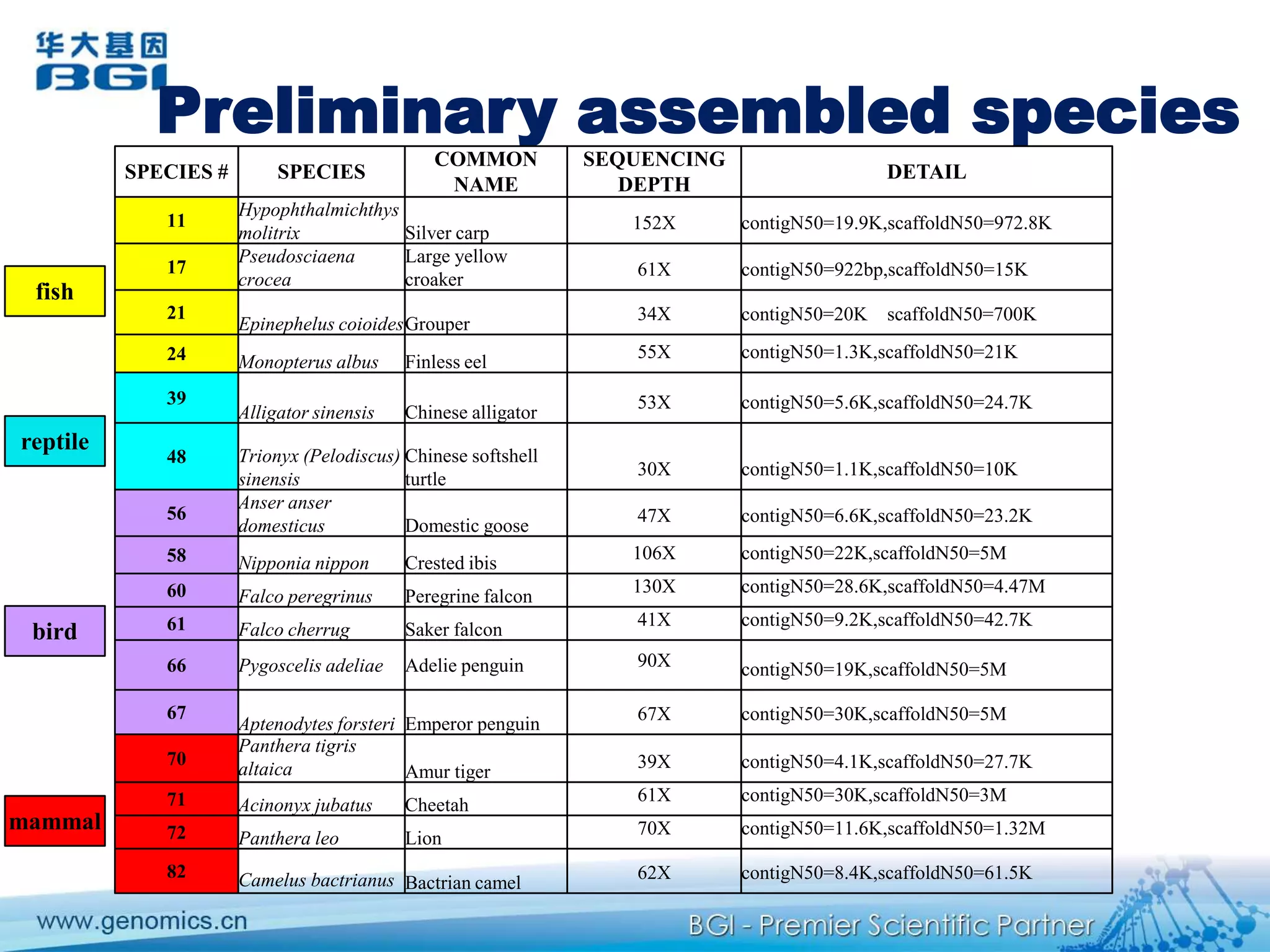




This document discusses the progresses and challenges of de novo genome assembly using next-generation sequencing data, including improvements made to error correction, contig construction, scaffolding, gap closure, and computational performance that have increased assembly quality and scalability; however, challenges still remain around resolving repeats and assembling heterozygous diploid genomes accurately.


































![[2013.10.29] albertsen genomics metagenomics](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131029070115-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)








































































