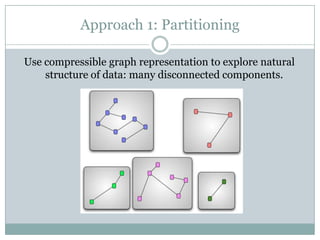
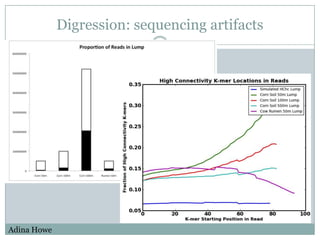
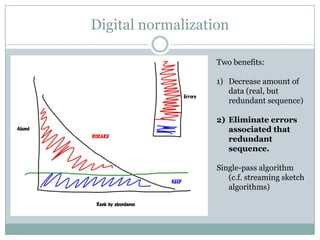
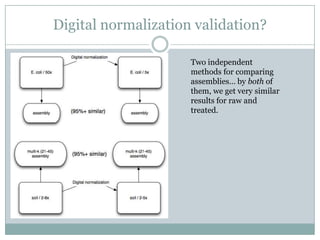
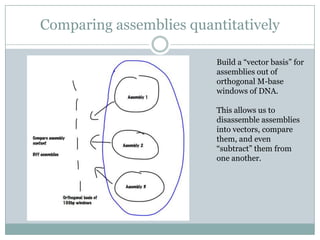
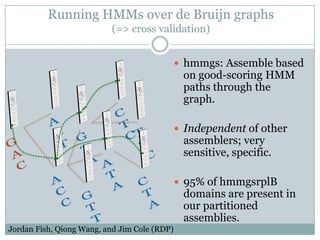
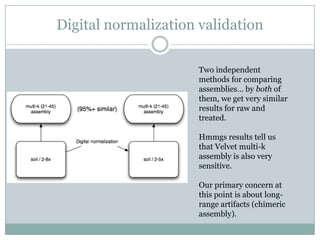
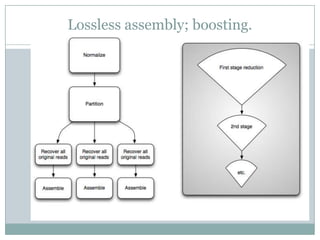
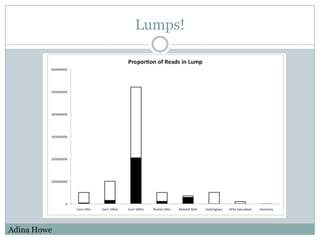
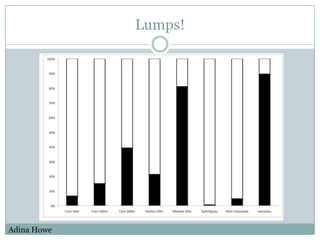
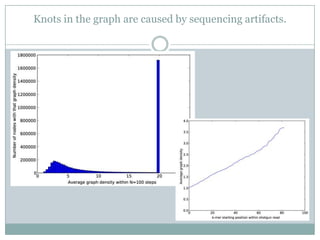
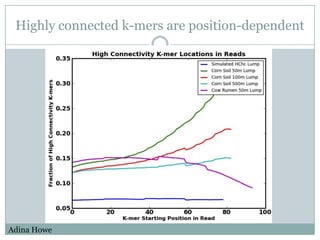
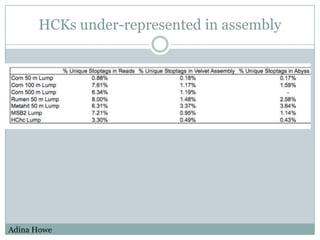
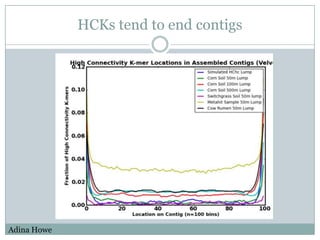
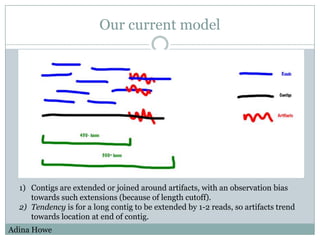
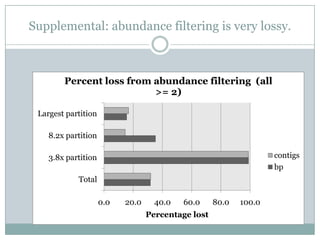
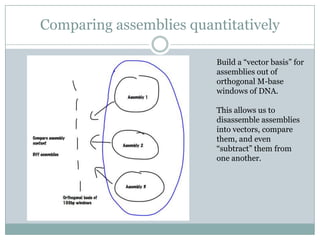
The document discusses scaling metagenome assembly to very large datasets. It proposes two approaches: 1) Partitioning the de Bruijn graph to divide the data into smaller, independent components that can be assembled in parallel. However, partitioning faces the same challenges of scaling to large datasets as other assemblers. 2) Digital normalization, which "squashes" redundant high coverage reads in a single pass to reduce the total data while preserving sequence content and gene coverage. Initial assemblies of field samples demonstrate the approaches can assemble petabase-scale datasets on a single compute node. Future work involves optimizing partitions, scaling implementations, and integrating other data types.