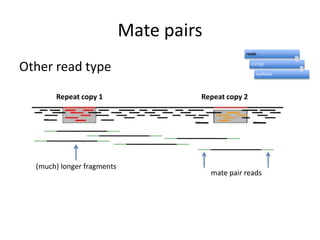
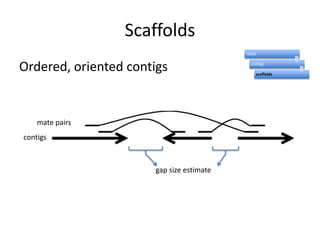
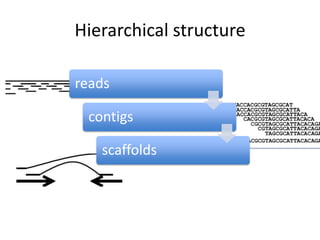
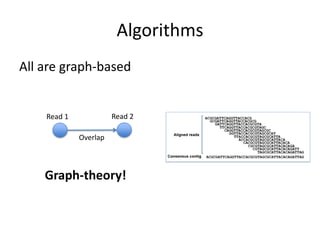
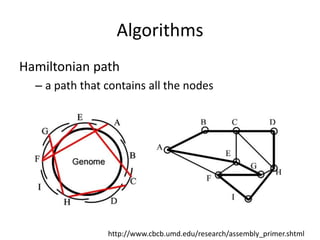
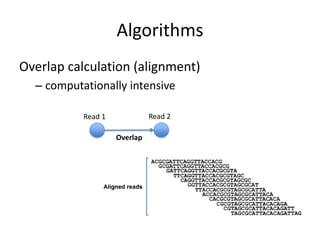
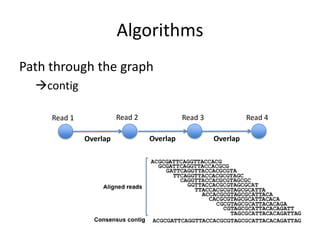
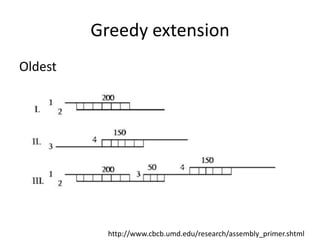
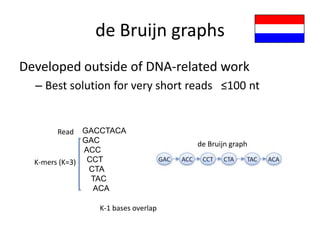
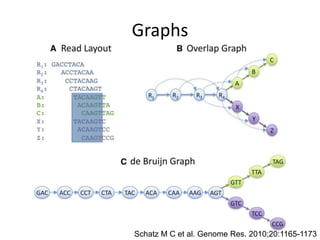
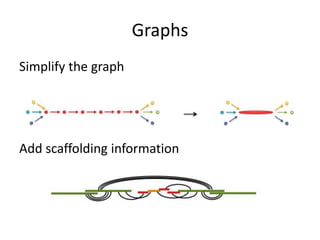
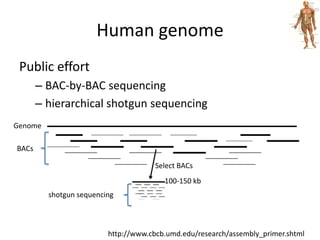
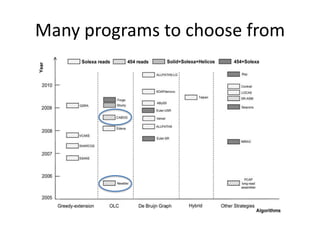
The document discusses the sequencing of large eukaryotic genomes, highlighting methods such as hierarchical shotgun sequencing and de Bruijn graphs. It details the specific case of the cod genome project, including the strategies employed, the assembly process, and challenges encountered. Additionally, it touches upon advancements in sequencing technology and bioinformatics for improved genome assembly.