Downloaded 346 times










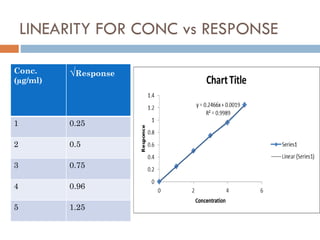









































This document discusses analytical method validation and cleaning validation in pharmaceutical manufacturing. It defines validation and outlines key parameters assessed in analytical method validation, including linearity, range, specificity, precision, accuracy, detection and quantitation limits, robustness, and system suitability. It also discusses objectives of cleaning validation, levels of cleaning, validation of cleaning processes and equipment, sampling methods, establishment of limits for residues, and documentation requirements. The overall purpose of validation is to demonstrate that processes can consistently produce products meeting specifications.



































































