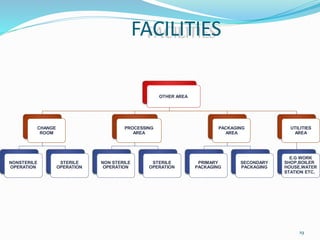
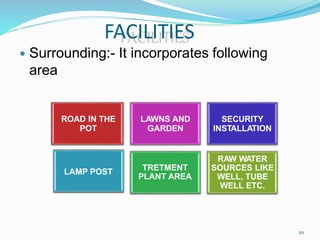
The document discusses cleaning validation in pharmaceutical manufacturing, emphasizing the importance of ensuring cleanliness to prevent contamination and cross-contamination of products. It outlines guidelines from the FDA on analytical methods related to cleaning validation, including specificity, sensitivity, accuracy, and precision. Additionally, it highlights the stages of developing cleaning validation protocols and the factors affecting the effectiveness of cleaning methods.














































































![Getting Started with Apache Spark: Big Data Made Simple [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/apachesparkgettingstarted-260203175547-8361bcc3-thumbnail.jpg?width=640&height=640&fit=bounds)











