Downloaded 2,684 times







































![Phase II
• Drug studied for 1st time in pts. with target disease
• Main purpose Gather evidence that drug has
effects suggested by preclinical trials
• End points :-
1. Definitive end point (Measures drug effect
directly Pain relief [analgesic])
2. Surrogate end point (Predictive of the definitive
end point Reduction in tumor size[anticancer])](https://image.slidesharecdn.com/drugdiscoveryanddevelopment-141116003217-conversion-gate02/85/Drug-discovery-and-development-50-320.jpg)












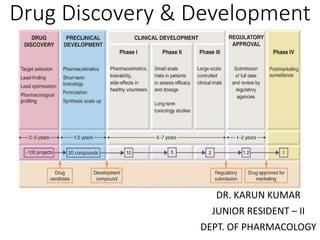
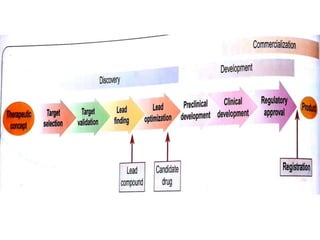
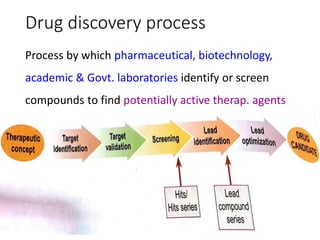
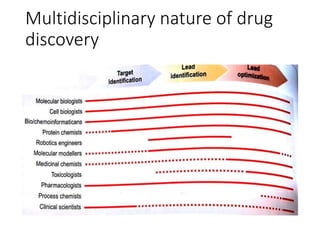
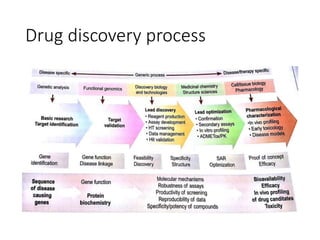
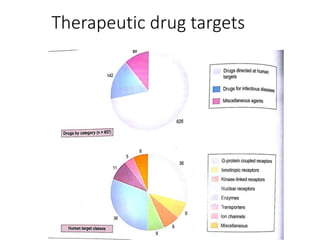
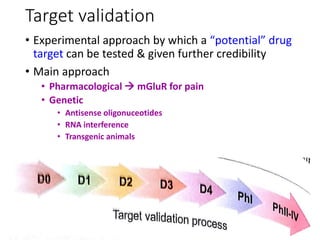
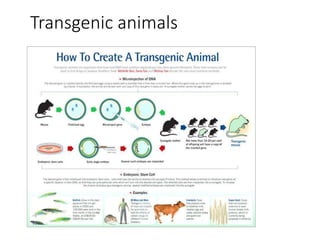
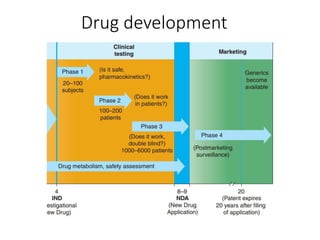
The document outlines the comprehensive process of drug discovery and development, which includes drug discovery (identifying potential therapeutic targets), drug development (preclinical studies and regulatory approval), and commercialization (marketing of the drug). It details various strategies for target identification, lead optimization, phases of clinical trials, and essential definitions and steps in preclinical and clinical development. Additionally, it emphasizes the multidisciplinary nature of drug discovery, involving genetics, pharmacokinetics, and animal models.


















![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)



![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)


















































































