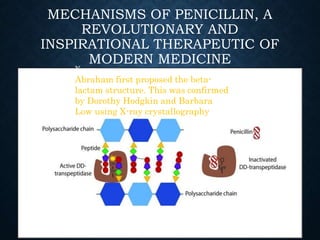
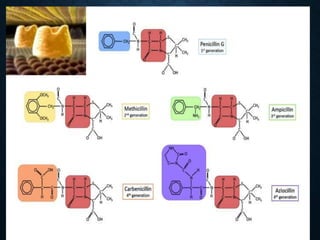
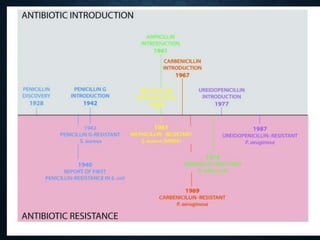
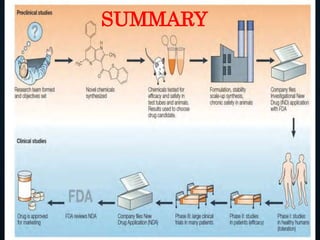
The document summarizes the key events in the discovery and development of penicillin. Alexander Fleming first discovered penicillin in 1928 after noticing bacteria-killing properties of the Penicillium mold in one of his petri dishes. However, he was not able to purify or characterize penicillin at the time. In the 1940s, a team at Oxford led by Howard Florey and Ernst Chain managed to purify and mass produce penicillin, paving the way for clinical trials. The first successful human trials demonstrated penicillin's ability to cure bacterial infections. By the mid-1940s, large-scale production was established. Fleming, Florey and Chain received the 1945 Nobel Prize for their discoveries. Over time, resistance












































![THE FIRST TRIALS IN HUMANS
• 1st patient – Cancer patient Fever and rigor d/t
impurities
• Edward Abraham – pyrogens purified
• 2nd patient - policeman at the Radcliffe Infirmary
who had severe staphylococcal and streptococcal
infection [5].
• Repeated intravenous injections of penicillin over 5
days had a profound effect on his recovery
Re extraction
from patients
urine
D/T short
supplyrelapse
and death](https://image.slidesharecdn.com/drugdevelopmentprocess-190528042628/85/Drug-development-process-57-320.jpg)