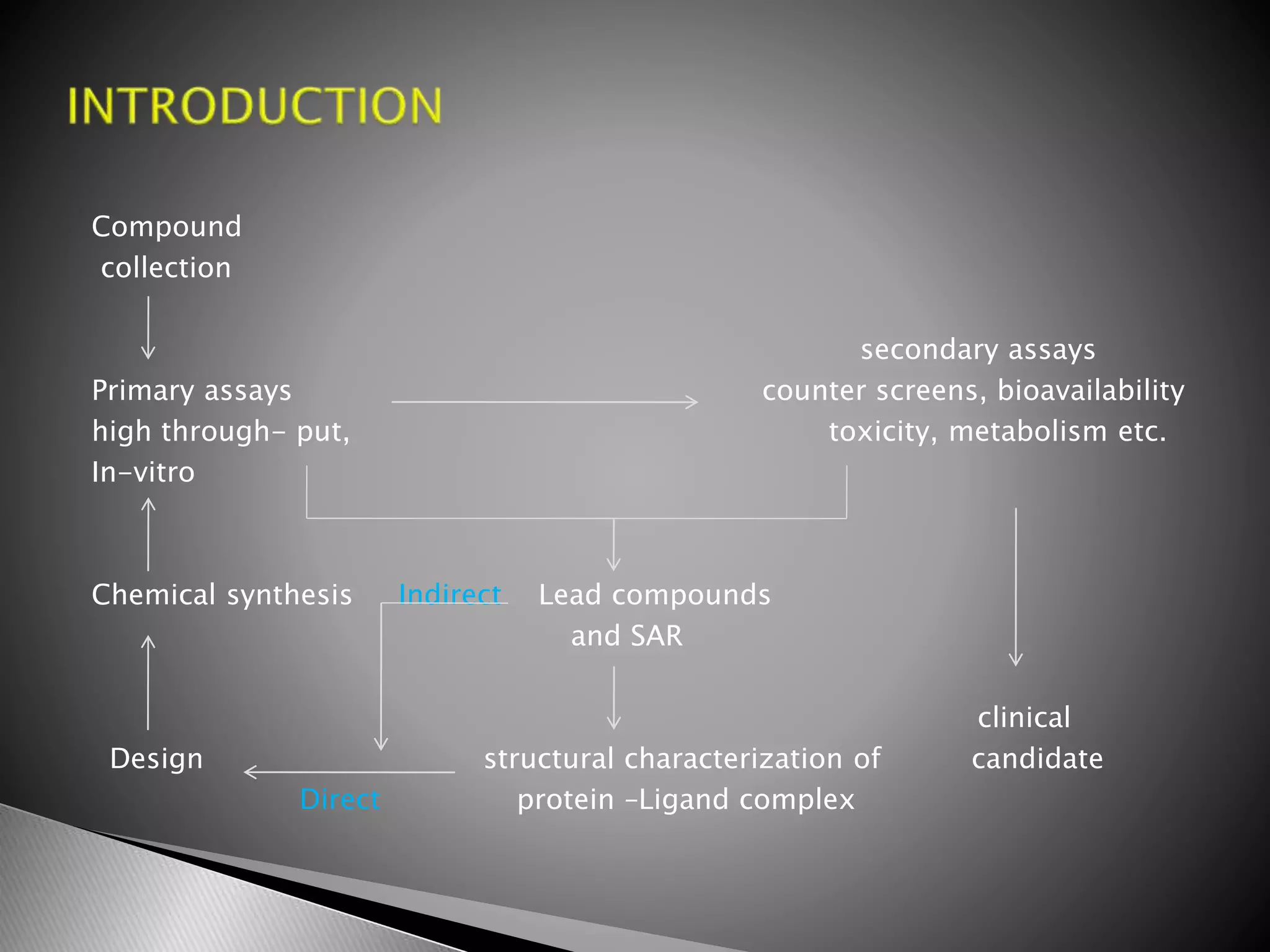
The document outlines the drug development process, detailing stages such as discovery, preclinical, and clinical trials, and the submission of investigational new drug applications (IND) to the FDA. It explains the methodology behind drug testing for safety and efficacy, including toxicology, pharmacokinetics, and the optimization of lead compounds. Additionally, it highlights the importance of clinical trials in bringing new treatments to market, along with their various types and the requirements needed to initiate them.


































![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)



















































