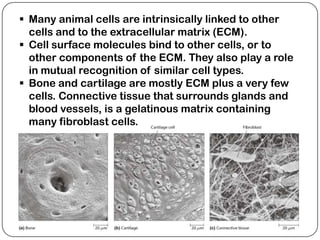
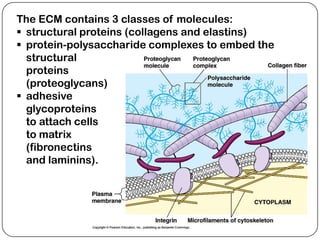
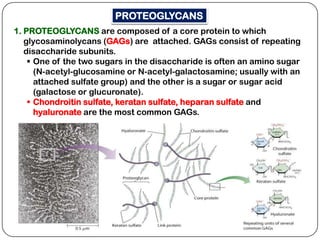
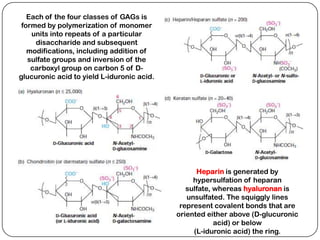
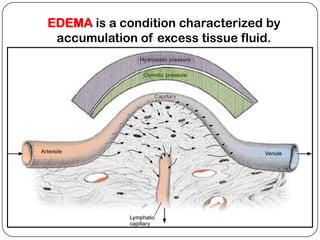
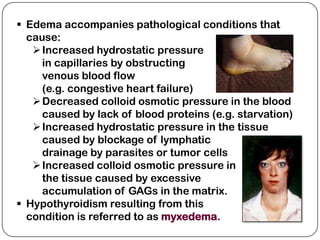
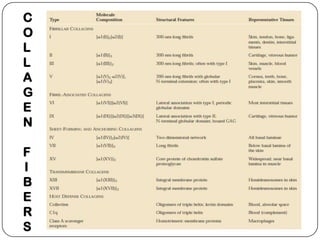
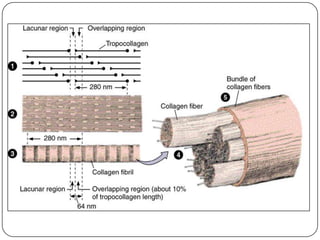
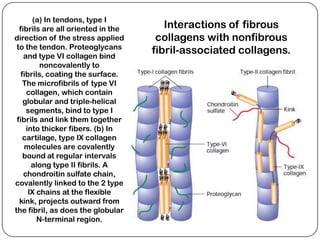
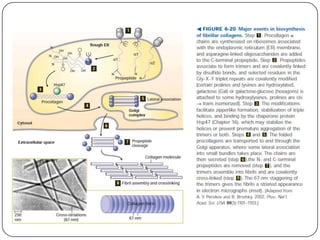
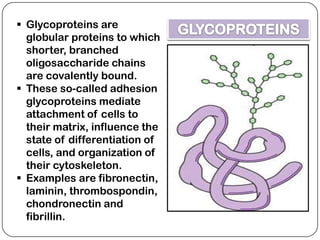
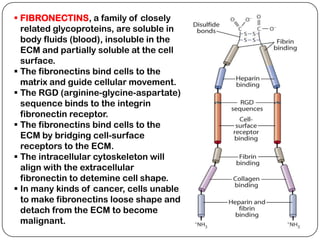
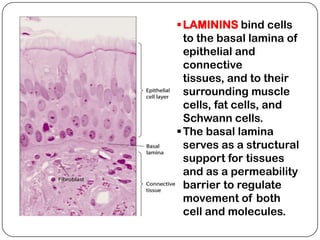
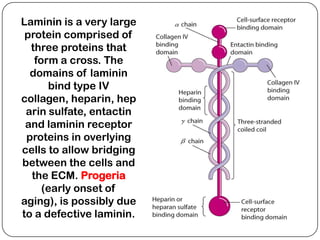
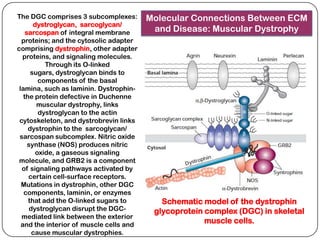
The document summarizes key components of the extracellular matrix (ECM). It describes three main classes of ECM molecules: structural proteins like collagen and elastin that provide structure, proteoglycans that embed the structural proteins, and adhesive glycoproteins like fibronectin and laminin that attach cells to the matrix. It provides details on the composition, structure and function of proteoglycans, collagen, elastic fibers, reticular fibers and adhesive glycoproteins. It also discusses how defects in ECM synthesis can lead to diseases and conditions like muscular dystrophy.



















![Extracellular matrix [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/extracellularmatrixautosaved-210313065413-thumbnail.jpg?width=640&height=640&fit=bounds)

































































































