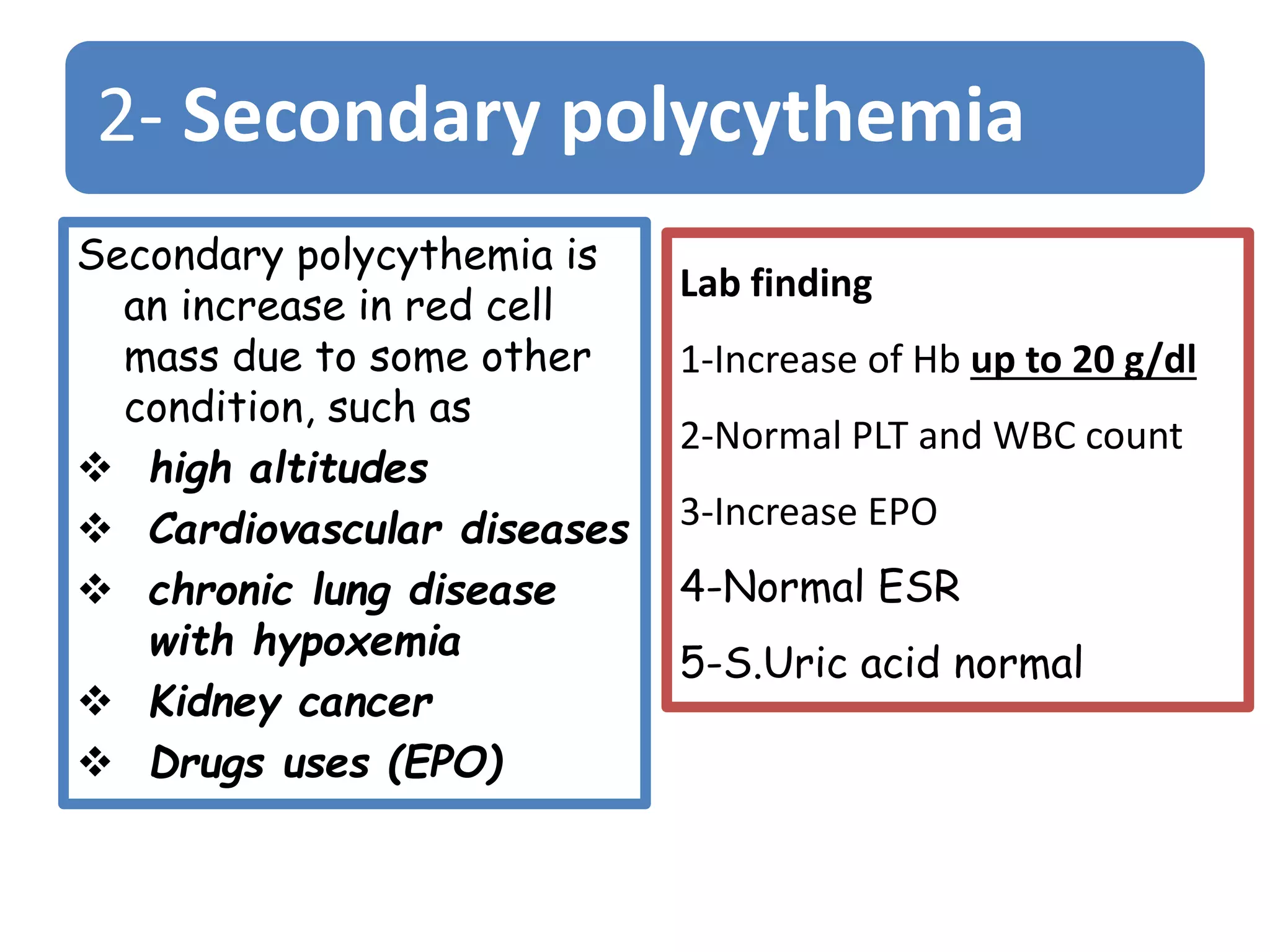
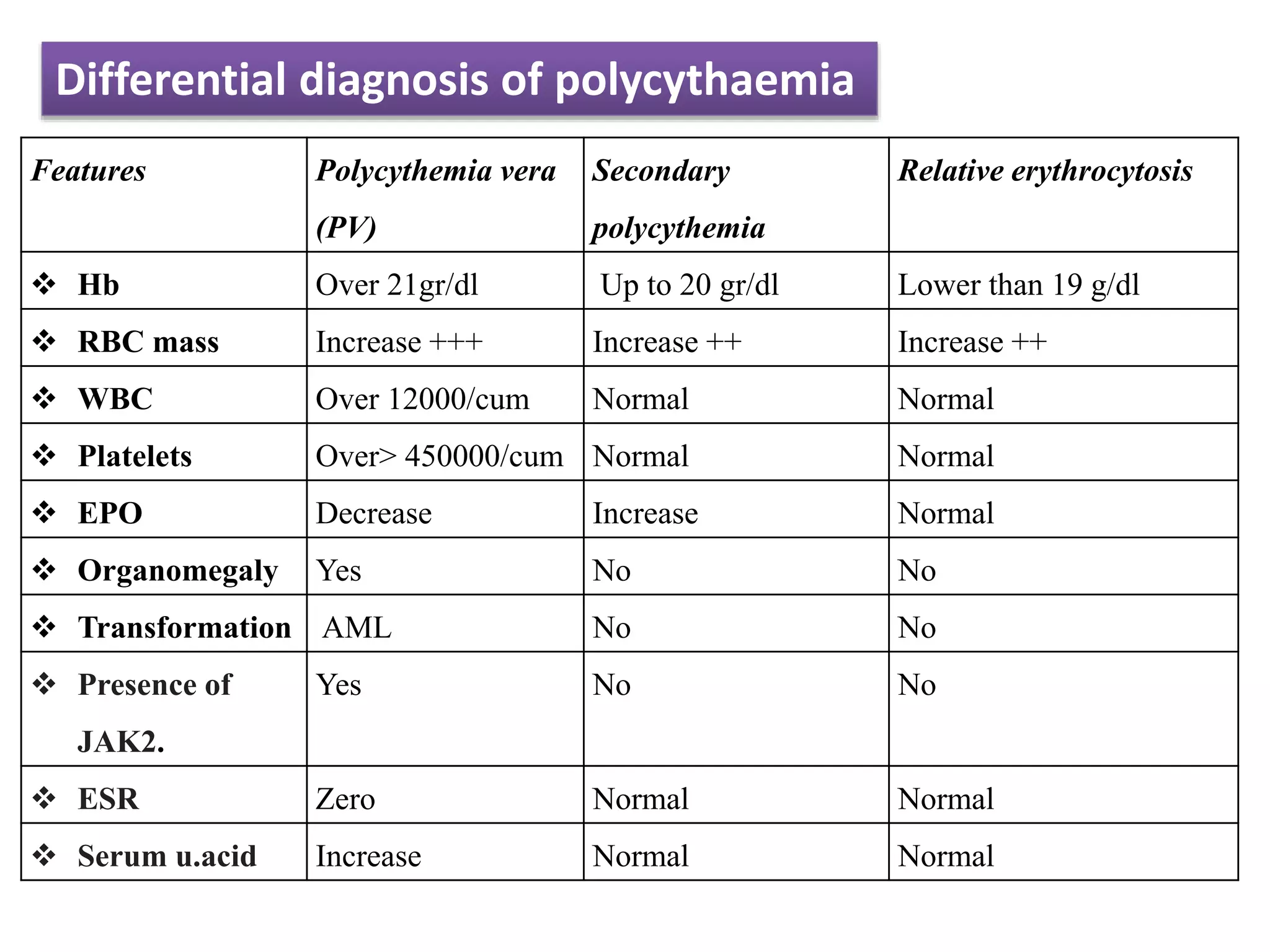
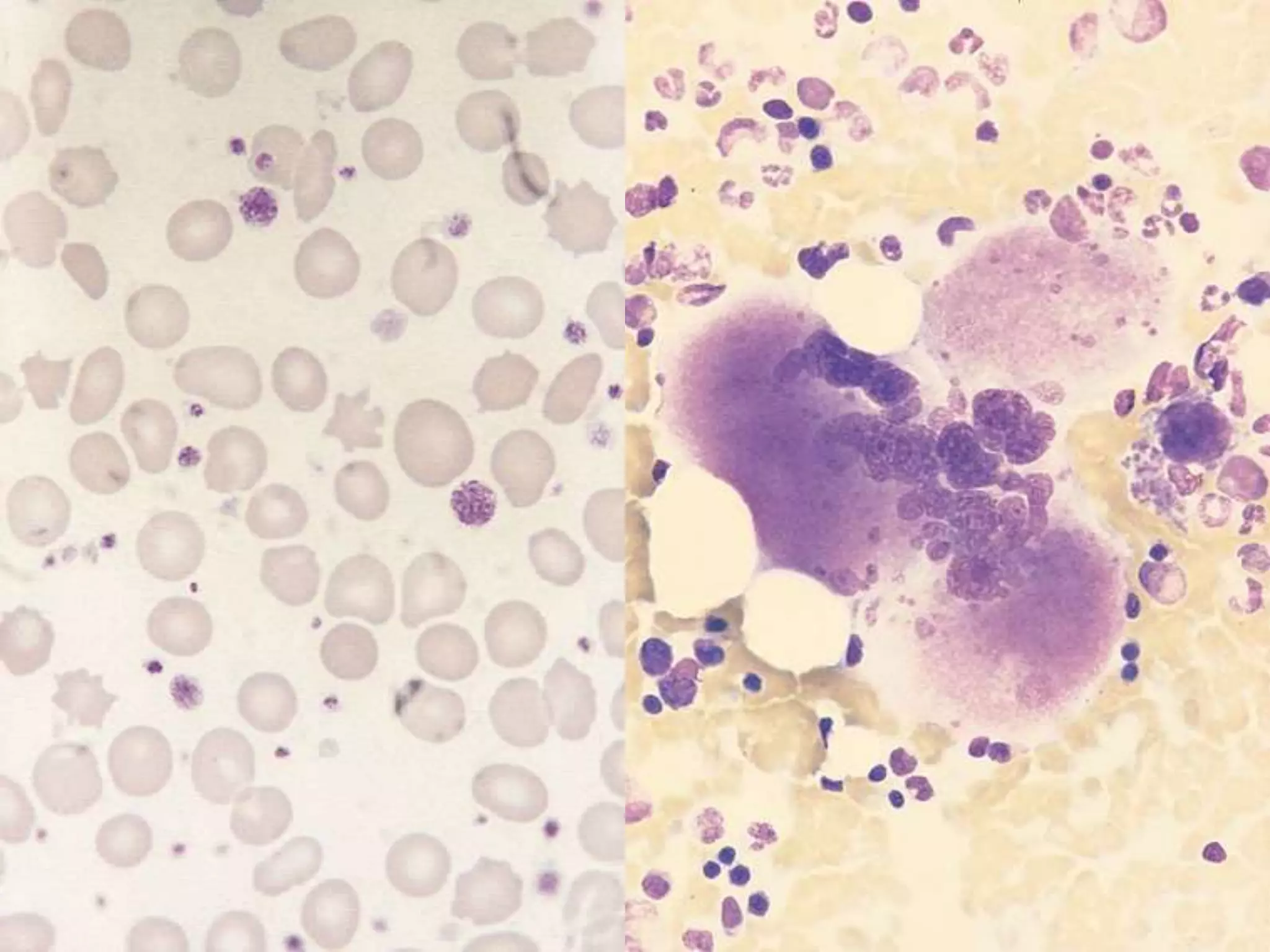
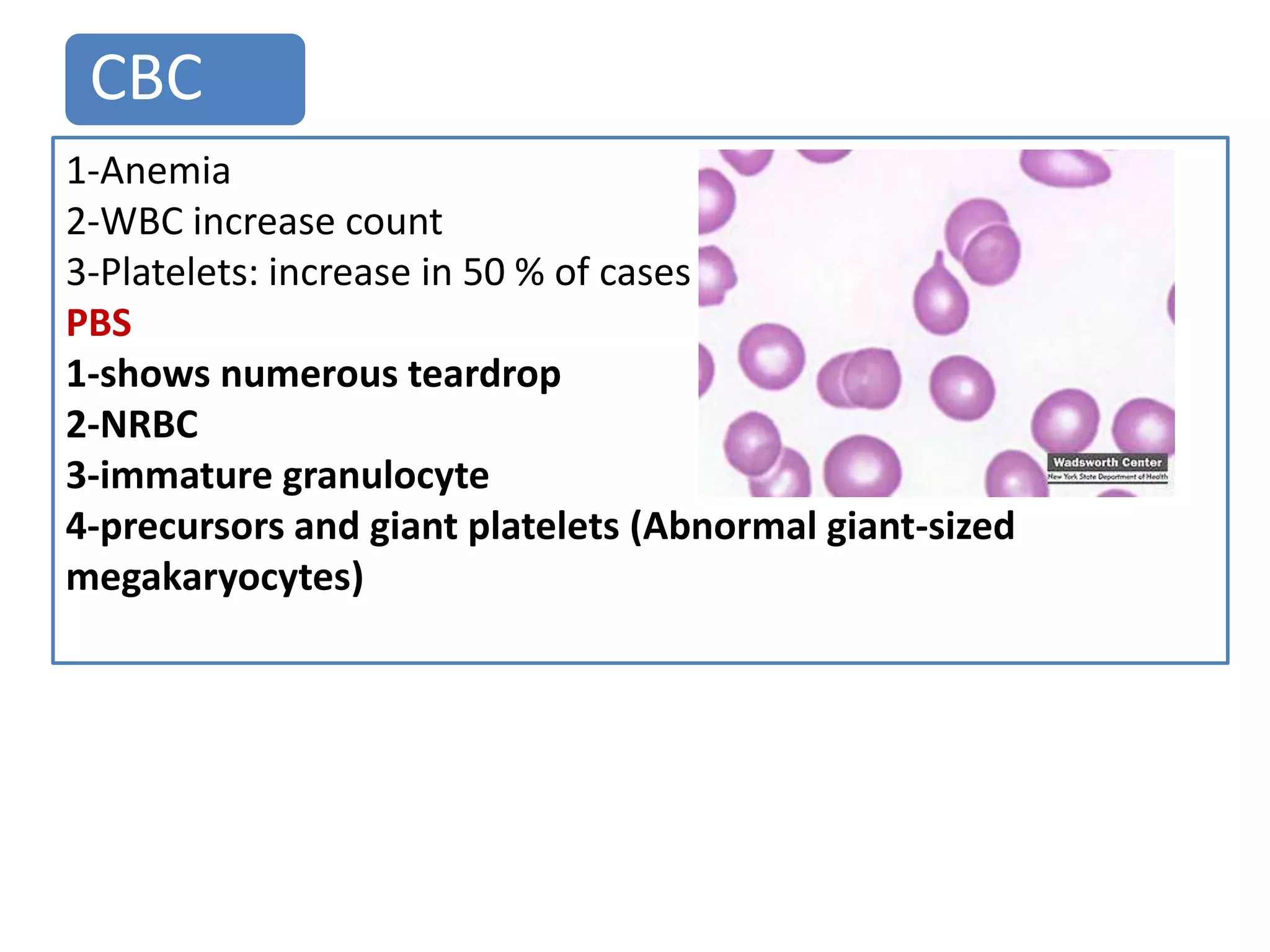
Polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis are related myeloproliferative neoplasms. PV is characterized by excessive production of all blood cell lines including red blood cells, white blood cells, and platelets independent of erythropoietin. The JAK2 mutation is present in almost all PV cases. ET involves increased megakaryocytes and platelet counts without elevated thrombopoietin. Myelofibrosis features abnormal stem cells leading to bone marrow fibrosis, extramedullary hematopoiesis, and a leukoerythroblastic blood smear.