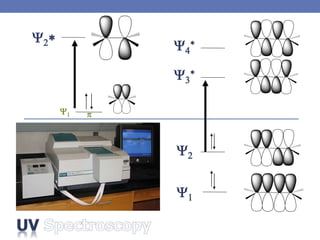

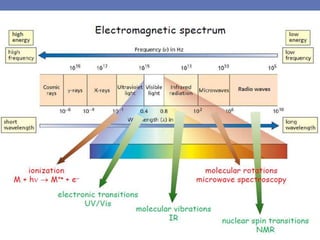
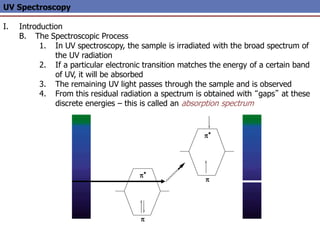

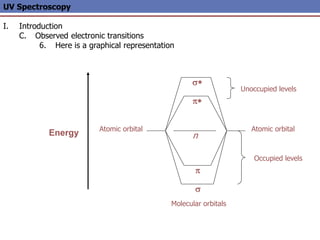
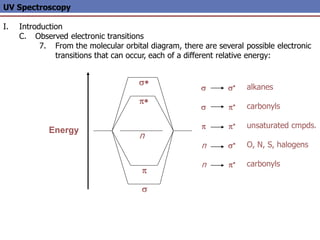



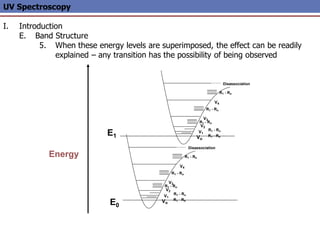
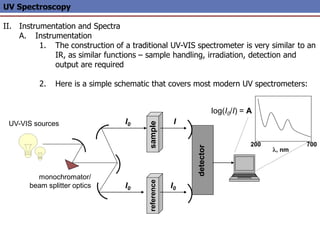

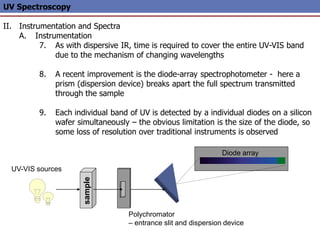






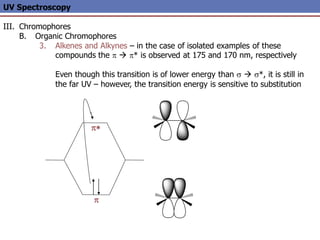

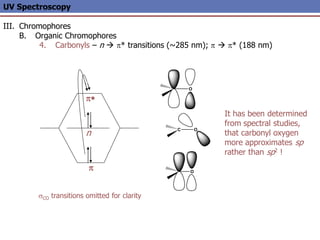

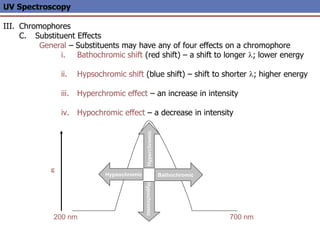

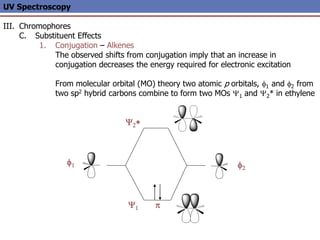
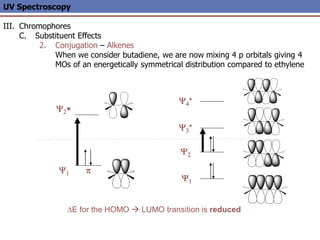
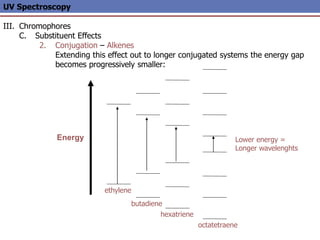
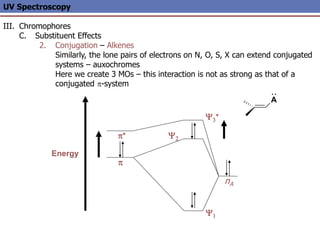
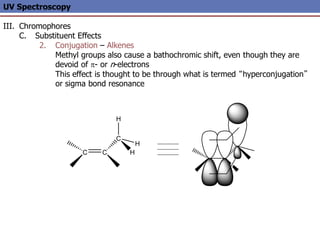
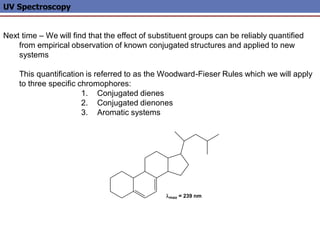





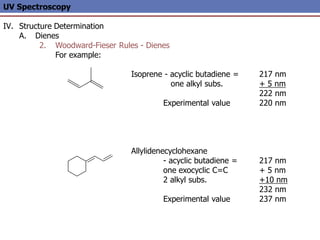
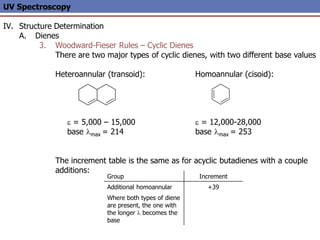
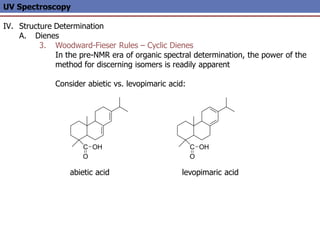

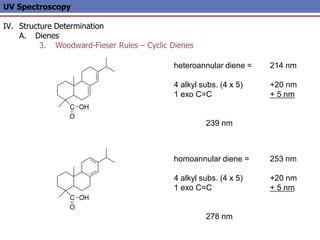
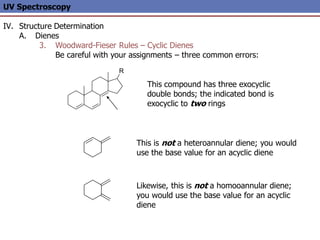
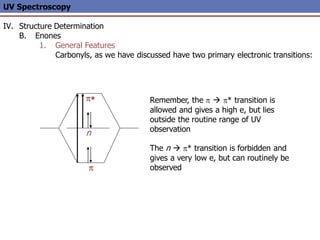


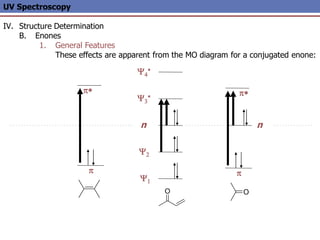
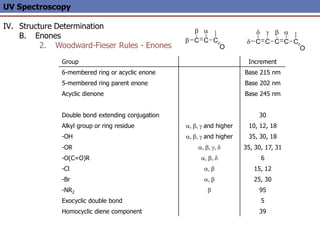
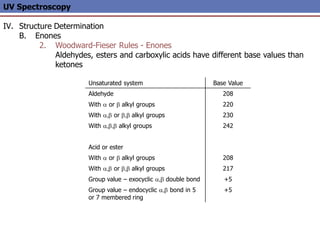
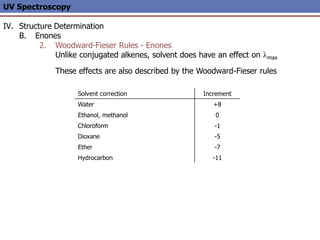


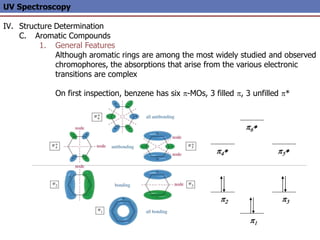



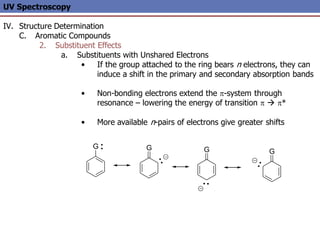
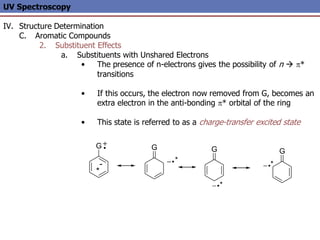
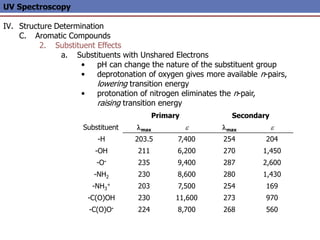
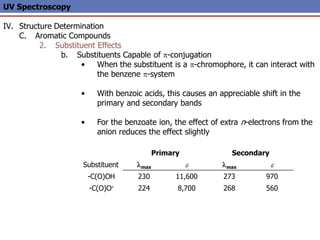

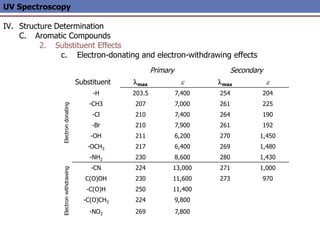


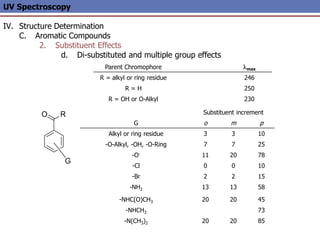
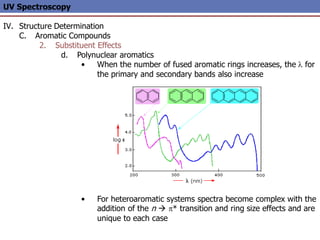


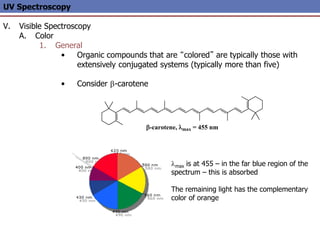
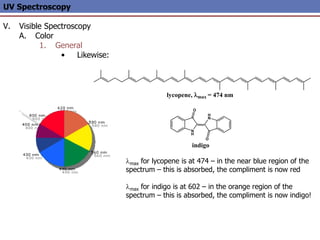
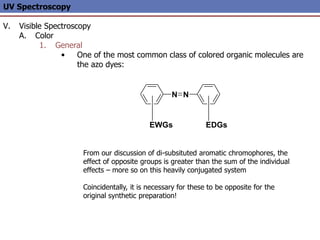
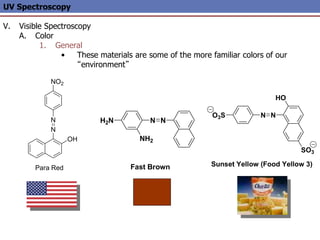
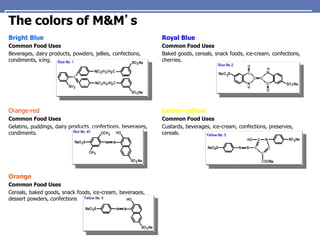
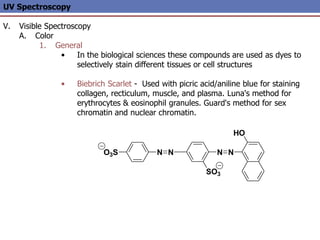
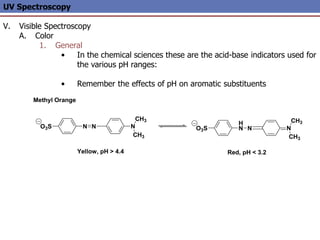


The document provides an extensive overview of UV spectroscopy, detailing the electronic energy transitions that occur in different molecular orbitals when samples are exposed to ultraviolet radiation. It explains the spectroscopic process, instrumentation, and the principles governing observed transitions and selection rules, along with the significance of chromophores and the effects of substituents on absorption characteristics. The practical applications of UV spectroscopy in combination with other analytical methods and its utility in laboratory settings, such as HPLC, are also discussed.























































































