Downloaded 358 times


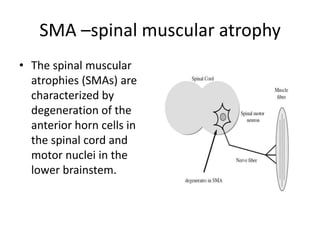


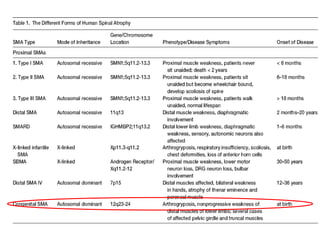
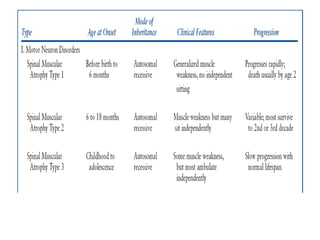
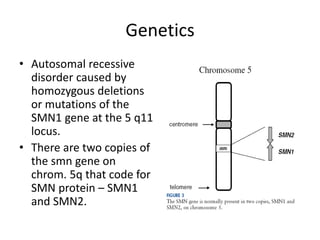

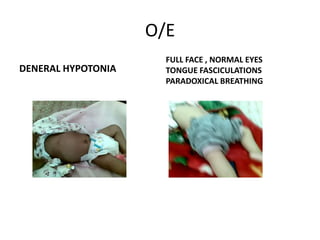
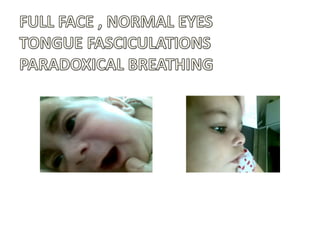
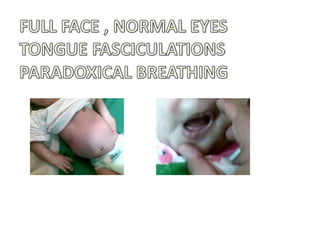
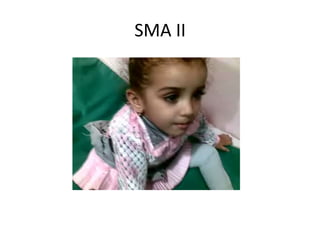










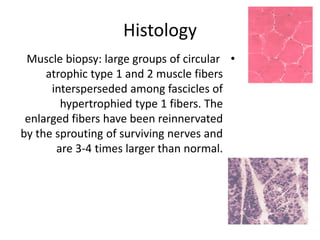
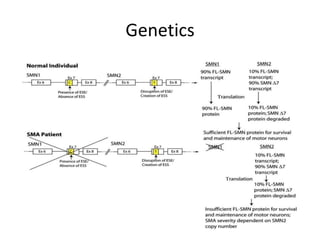
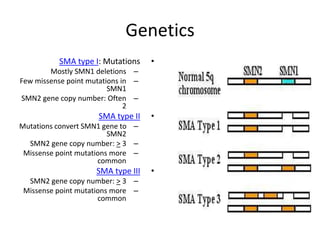
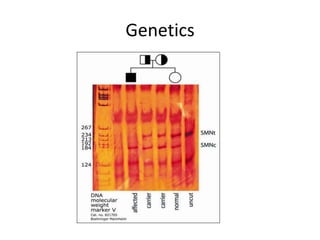




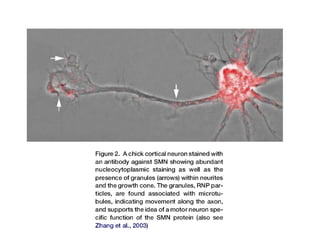

1. Spinal muscular atrophy (SMA) is caused by degeneration of motor neurons in the spinal cord and brainstem due to a defect in the SMN1 gene resulting in low levels of the SMN protein. 2. SMA is classified into five types based on age of onset and severity of symptoms - from very severe Type 1 to milder adult-onset Type 4. 3. Diagnosis involves family history, physical exam, genetic testing, and other tests like EMG; there is currently no cure but supportive care can help manage symptoms.































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)











