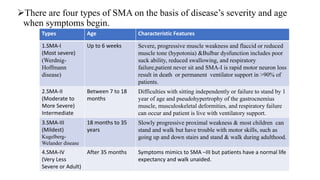
This document provides information about therapeutic options for spinal muscular atrophy (SMA). It first defines SMA as a genetic disorder that causes degeneration of motor neurons in the spinal cord, resulting in muscle weakness. It then discusses the four main types of SMA based on severity and age of onset. The causes and diagnosis of SMA are also outlined. The therapeutic options section summarizes gene replacement therapies like nusinersen and onasemnogene abeparovec, which aim to increase SMN protein levels. It also mentions the small molecule risdiplam and ongoing clinical trials evaluating these treatment approaches.


































![References
• Werdnig G: Zwei frühinfantile hereditäre Fälle von progressive Muskelatrophie unter dem Bilde der Dystrophie, aber auf neurotischer
Grundlage [Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis; in
German]. Arch Psychiatr Nervenkr 1891, 22:437-480.
• Hoffmann J: U” ber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis [On chronic spinal muscular atrophy in
childhood, with a familial basis; in German]. Dtsch Z Nervenheilkd 1893, 3:427-470
• Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D,
Dubowitz V, Zerres K, Hausmanowa-Petrusewicz I, Ott J, Munsat TL, Gilliam TC: Genetic mapping of chronic childhood-onset spinal
muscular atrophy to chromosome 5q11.2-13.3. Nature 1990, 344:540-41.
• Spinraza (nusinersen) [package insert]. Cambridge, MA: Biogen Inc. 2016 December.
• Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M.
Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897.
• Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al.
Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet Lond. Engl.
2016, 388, 3017–3026
• Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-Dose Gene-Replacement Therapy for Spinal
Muscular Atrophy. N Engl J Med. 2017 Nov 2. 377 (18):1713-1722.
• Al-Zaidy S, Pickard AS, Kotha K, Alfano LN, Lowes L, Paul G, et al. Health outcomes in spinal muscular atrophy type 1 following
AVXS-101 gene replacement therapy. Pediatr Pulmonol. 2019 Feb. 54 (2):179-185.
• Novartis. AveXis data reinforce effectiveness of Zolgensma in treating spinal muscular atrophy (SMA) Type 1. 2019 Apr 16.](https://image.slidesharecdn.com/spinalmuscularatrophy-201107090832/85/Spinal-muscular-atrophy-35-320.jpg)
![• Servais L, Baranello G, Masson R, et al. FIREFISH Part 2: efficacy and safety of risdiplam (RG7916)
in infants with spinal muscular atrophy (SMA) (1302). Neurology. April 14, 2020;94
• Evrysdi (risdiplam) [package insert]. South San Francisco, CA: Genentech. August 2020.
• Hwee, D.T.; Kennedy, A.; Ryans, J.; Russell, A.J.; Jia, Z.; Hinken, A.C.; Morgans, D.J.; Malik, F.I.;
Jasper, J.R. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and
performance in the B6SJL-SOD1G93AALS mouse model. PLoS ONE 2014, 9, e96921.
• Rudnicki, S.A.; Andrews, J.A.; Malik, F.I. CY 5021 A phase 2, double-blind, randomized, placebo-
controlled, multiple-dose study of reldesemtiv 2 ascending-dose cohorts of patients with Spinal
Muscular Atrophy (SMA). In Proceedings of the Cure SMA 2018, Dallas, TX, USA, 16 June 2018.
• Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, et al. SMA CARNIVAL
TRIAL PART II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory
children with spinal muscular atrophy. PLoS One. 2011. 6(7):e21296.
• Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, et al. SMA CARNI-
VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in
spinal muscular atrophy. PLoS One. 2010 Aug 19. 5(8):e12140.
• Wadman RI, Bosboom WM, van der Pol WL, van den Berg LH, Wokke JH, Iannaccone ST, et al.
Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2012 Apr
18. 4:CD006282.](https://image.slidesharecdn.com/spinalmuscularatrophy-201107090832/85/Spinal-muscular-atrophy-36-320.jpg)































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

















