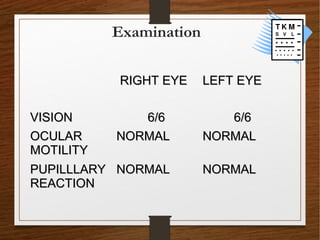
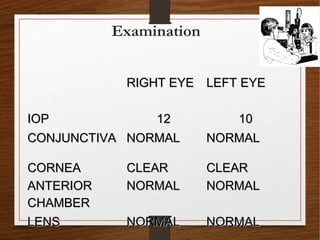
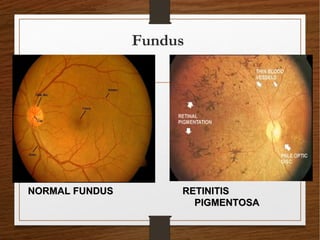
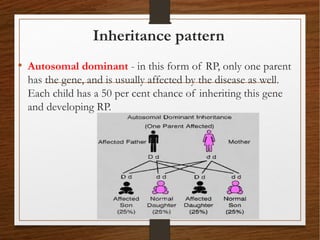
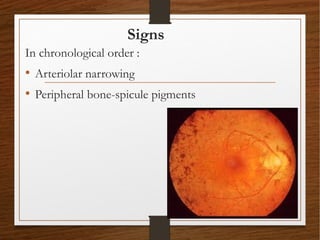
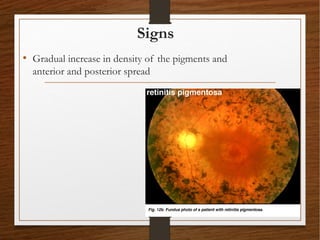
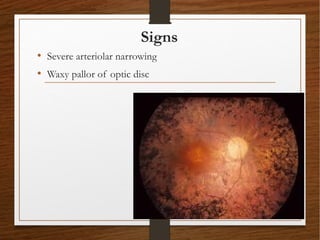
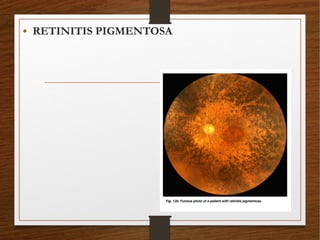
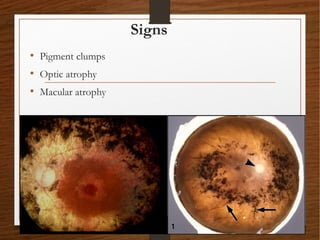
1) A 20-year-old boy presented with gradual decrease in night vision over 6 months. Examination revealed signs consistent with retinitis pigmentosa including bone-spicule pigmentation and waxy pale optic discs.
2) Retinitis pigmentosa is a hereditary retinal dystrophy where rod photoreceptor cells are initially and predominantly affected, followed by cone degeneration. It typically causes night blindness and progressive loss of peripheral vision.
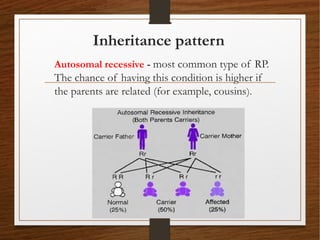
3) There is no cure for retinitis pigmentosa. Treatment aims to slow progression and improve quality of life through low vision aids, vitamin supplements, and emerging therapies like gene and retinal prosthesis. Prognosis depends on inheritance pattern, with