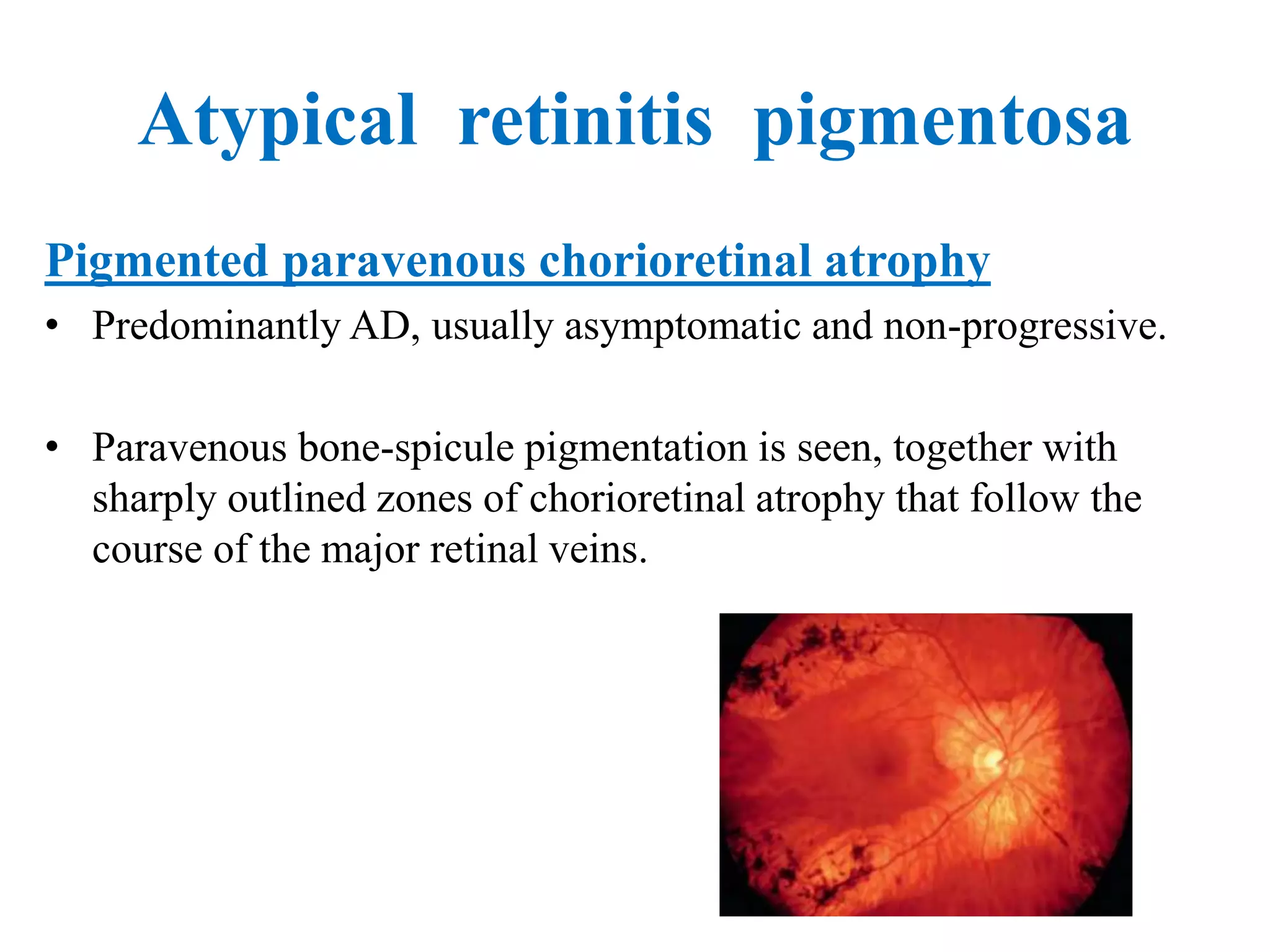
This document discusses hereditary retinal dystrophies. It begins by introducing retinal dystrophies and classifying them based on inheritance pattern and the pathological process affected. Several specific dystrophies are then discussed in more detail, including retinitis pigmentosa, Stargardt disease, and Best vitelliform macular dystrophy. For each, the document outlines clinical features, inheritance patterns, investigations, treatment approaches, and complications. Overall, the document provides an overview of hereditary retinal dystrophies with a focus on several common genetic subtypes.