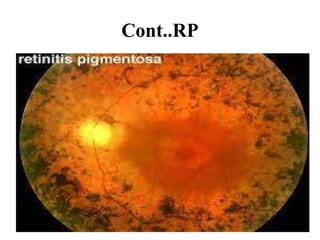
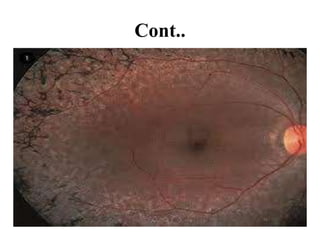
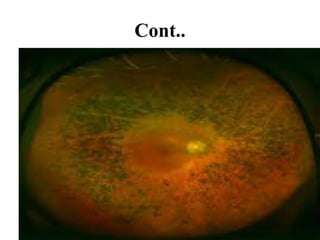
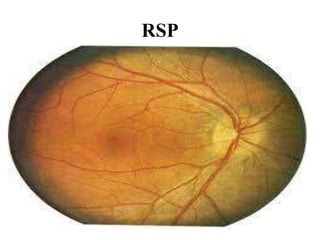
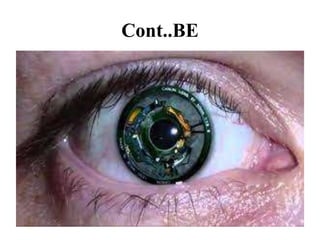
Retinitis pigmentosa is a rare inherited degenerative eye disease leading to severe vision impairment due to the loss of photoreceptors in the retina, initially affecting rods and subsequently cones. Symptoms include night blindness, poor visual acuity, and gradual loss of peripheral vision, ultimately resulting in blindness in adulthood. While there is currently no cure, management options include low vision aids, high-dose vitamin A, and potential advanced treatments like gene therapy.











































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)













![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)
