


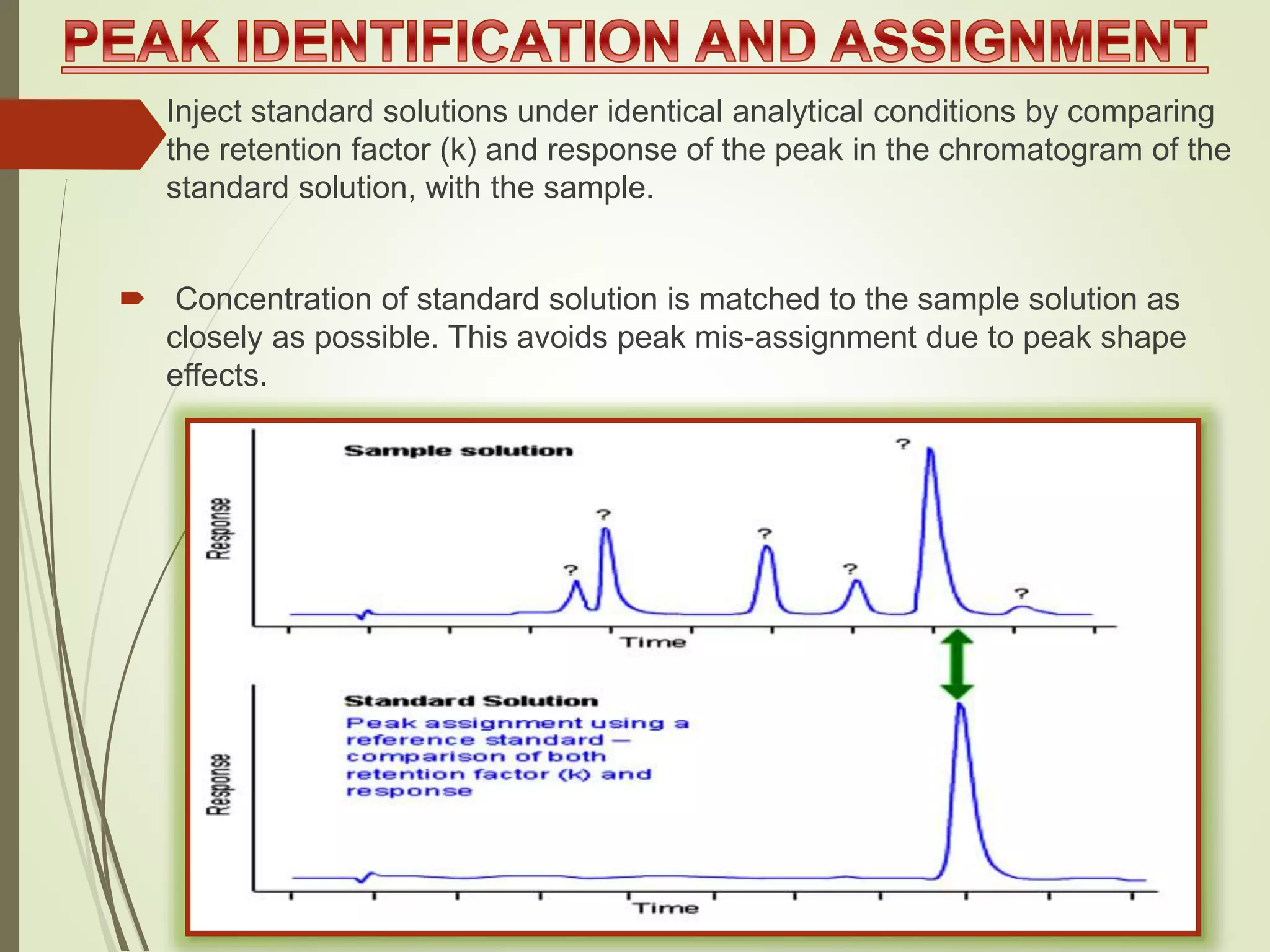


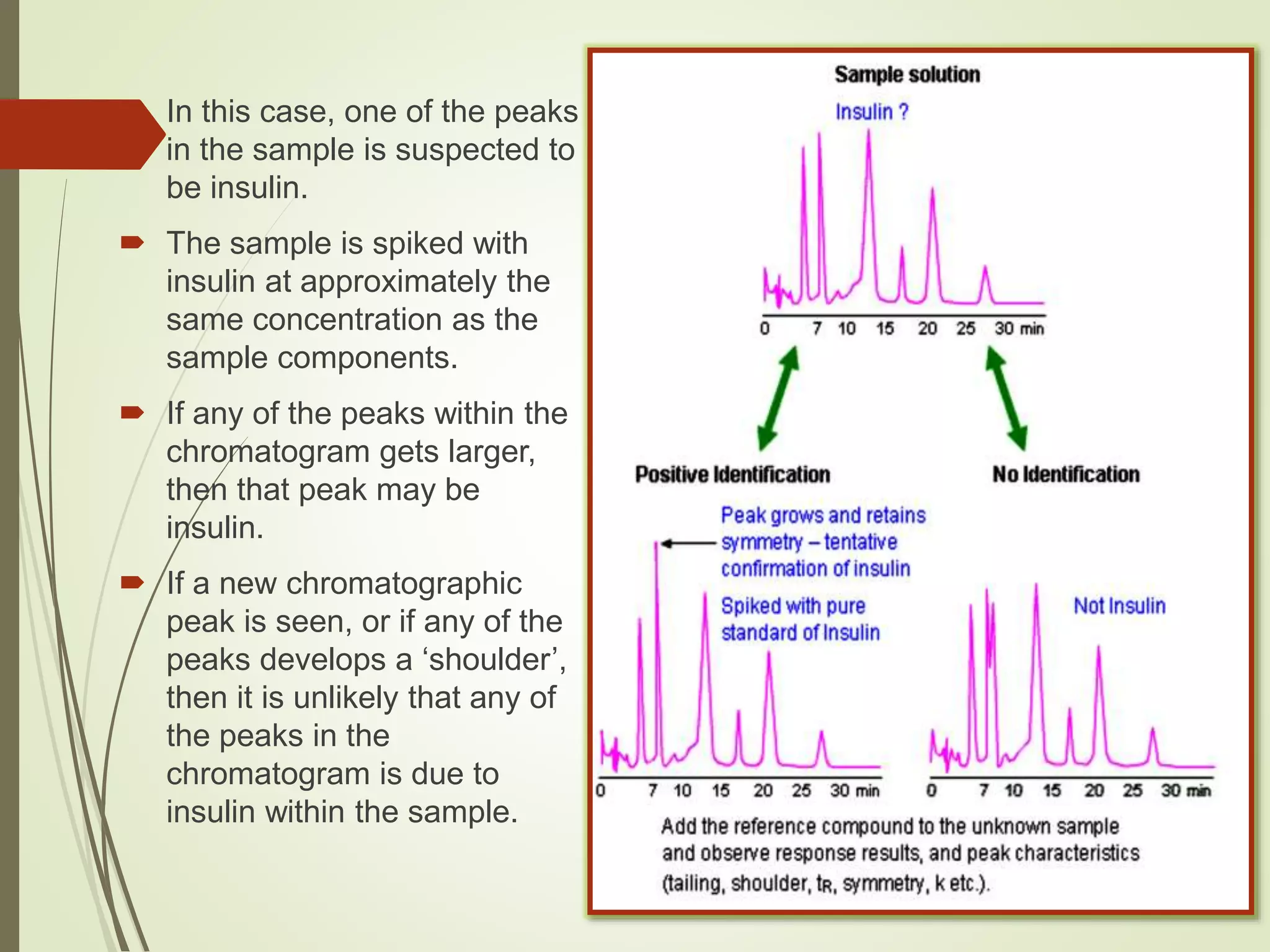
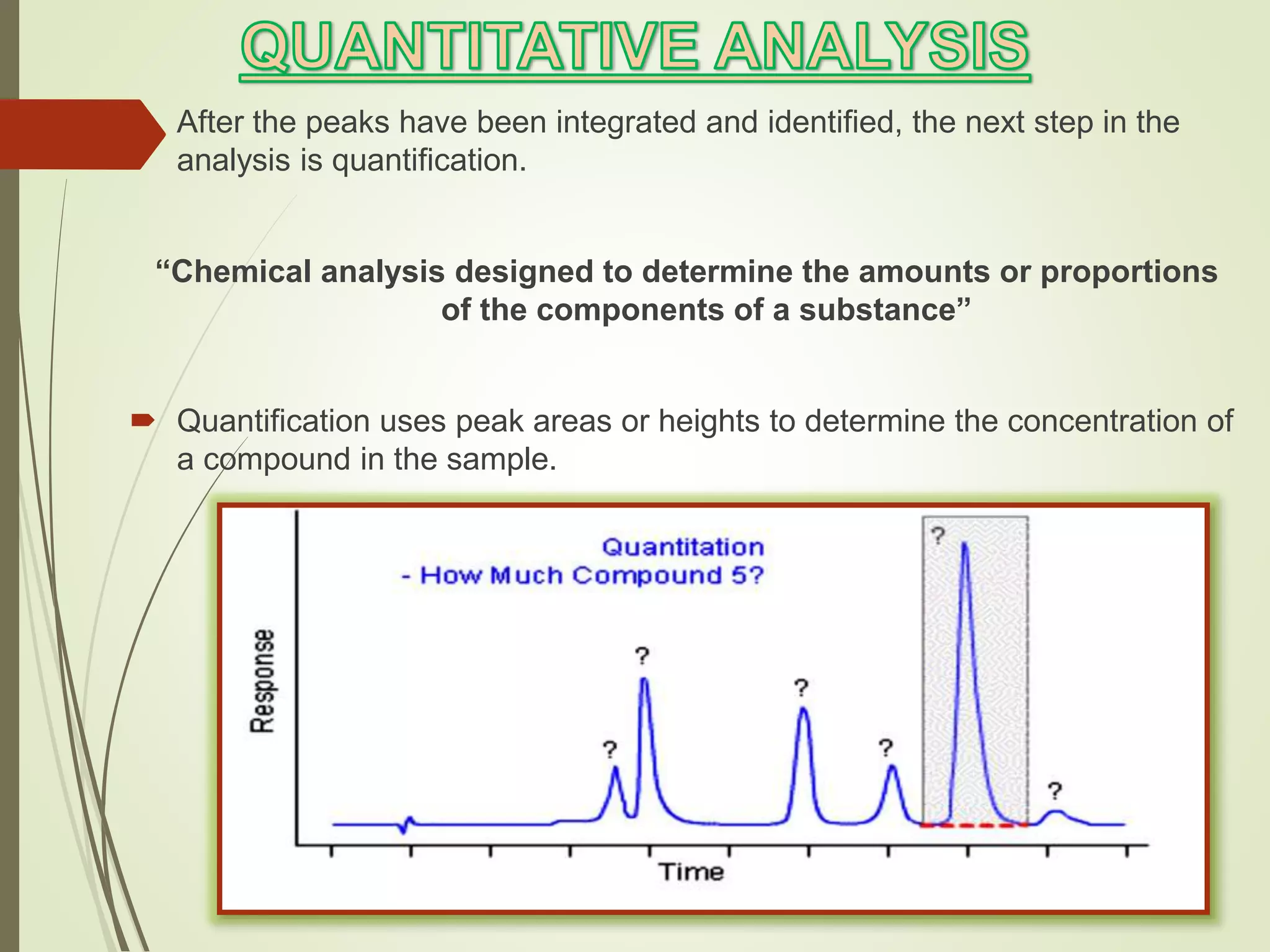

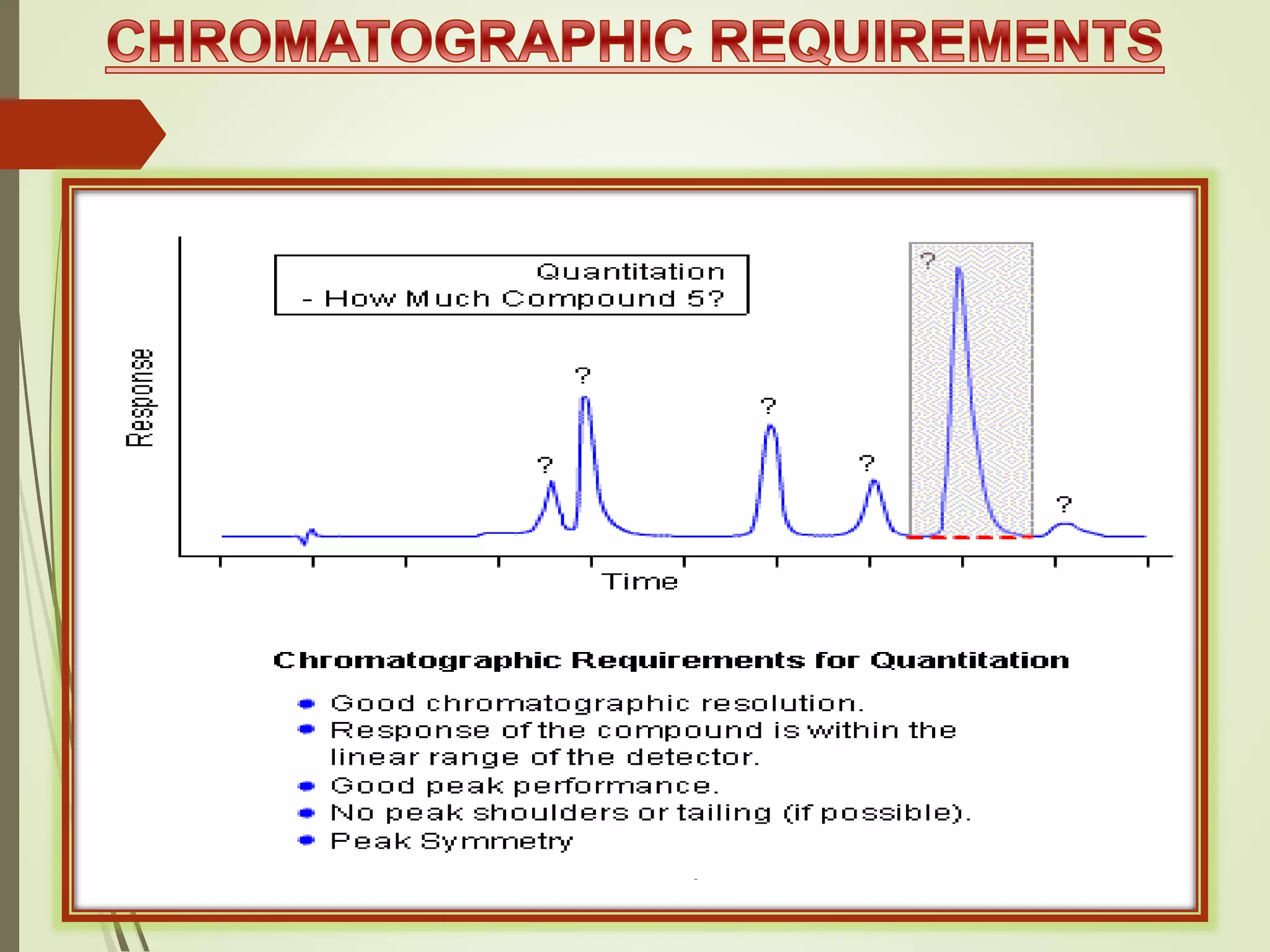
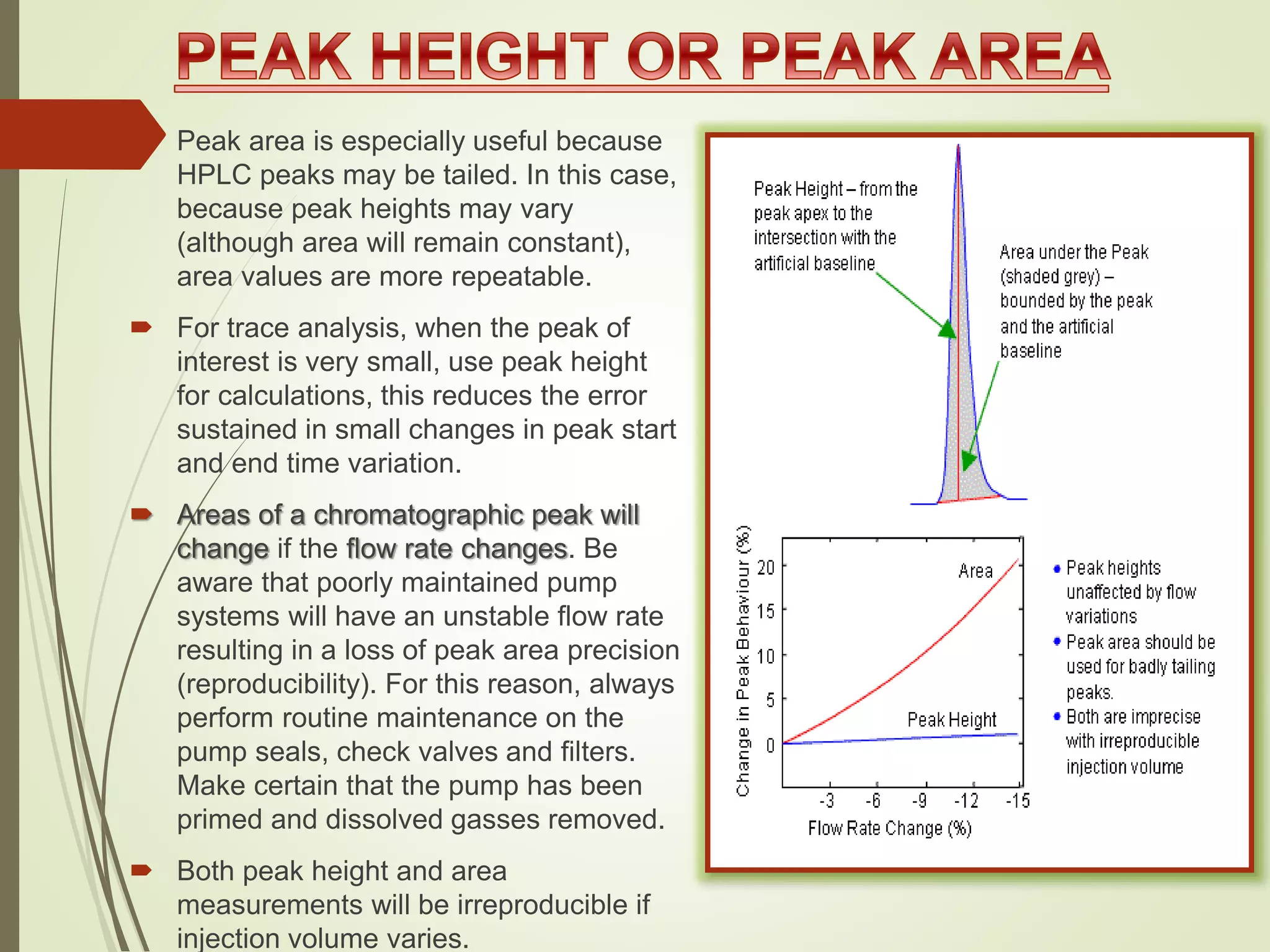

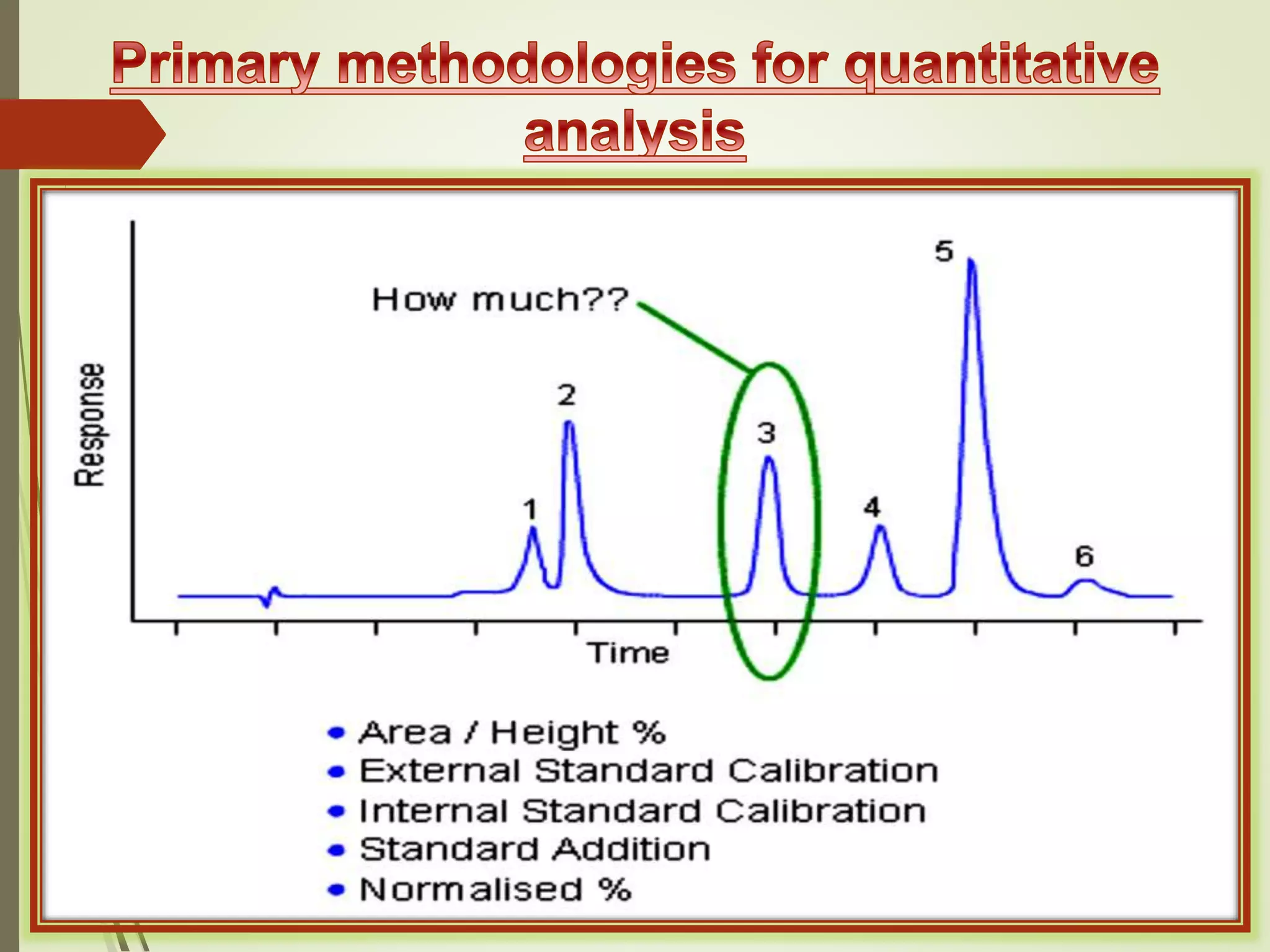
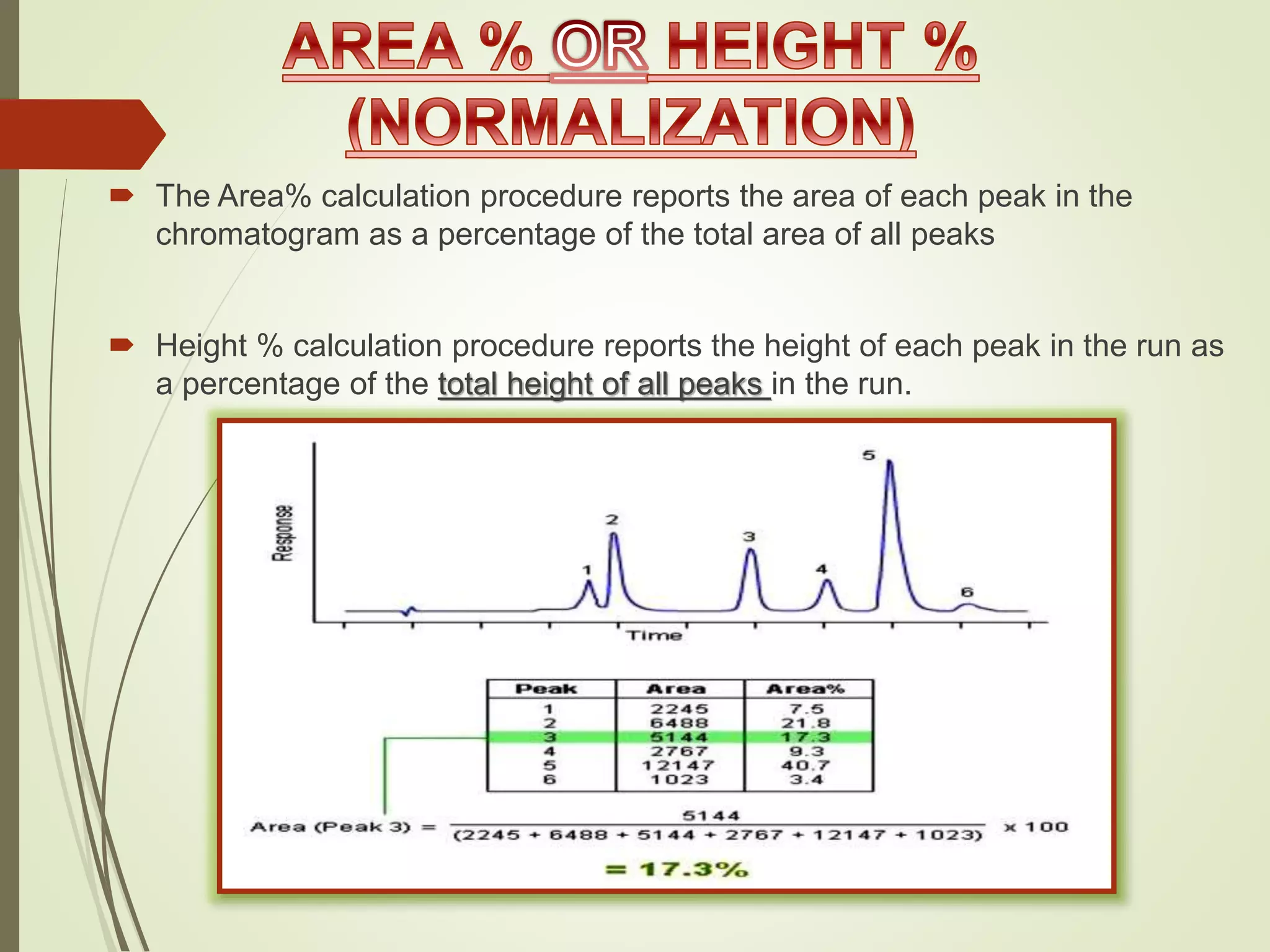





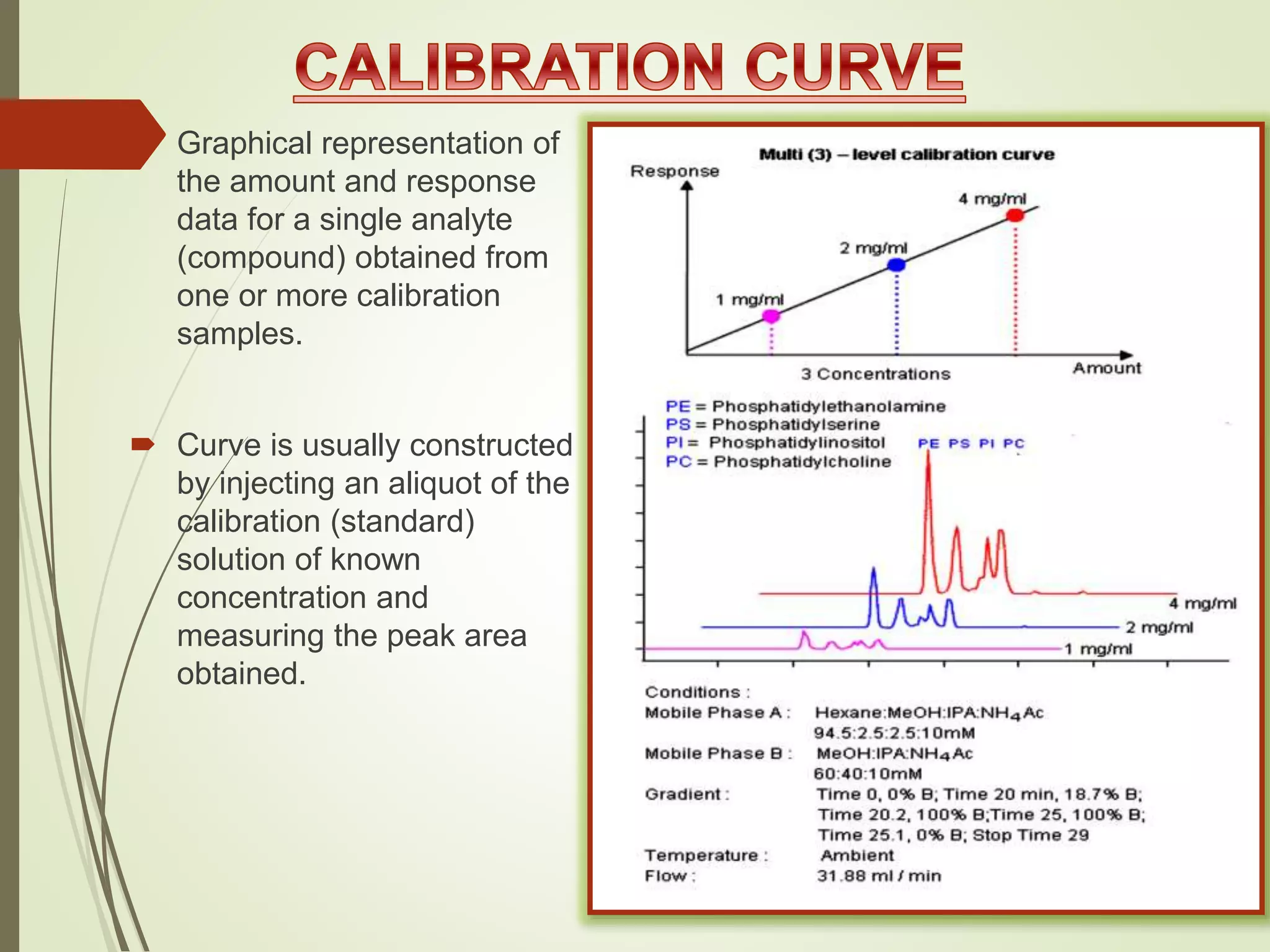
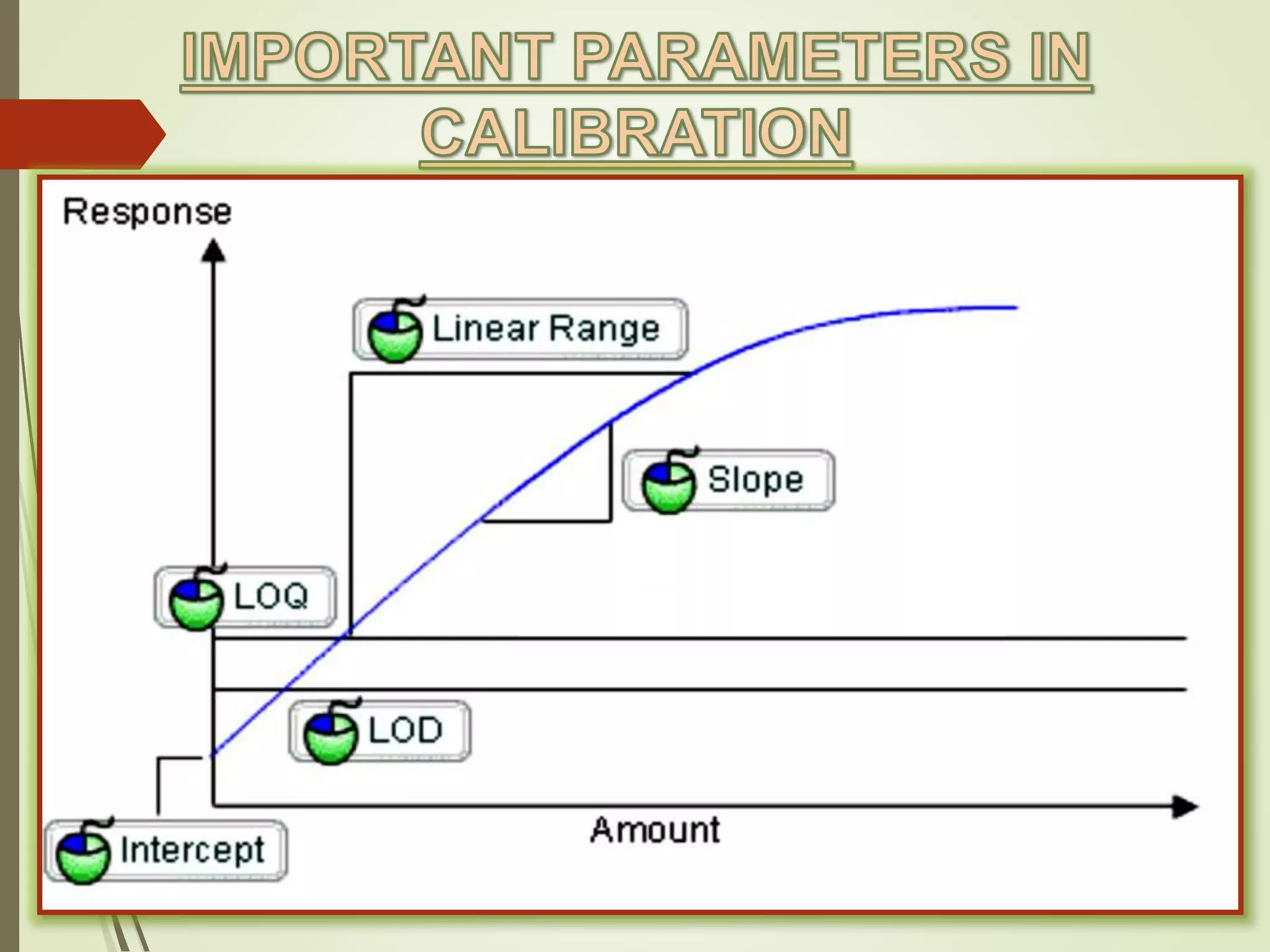

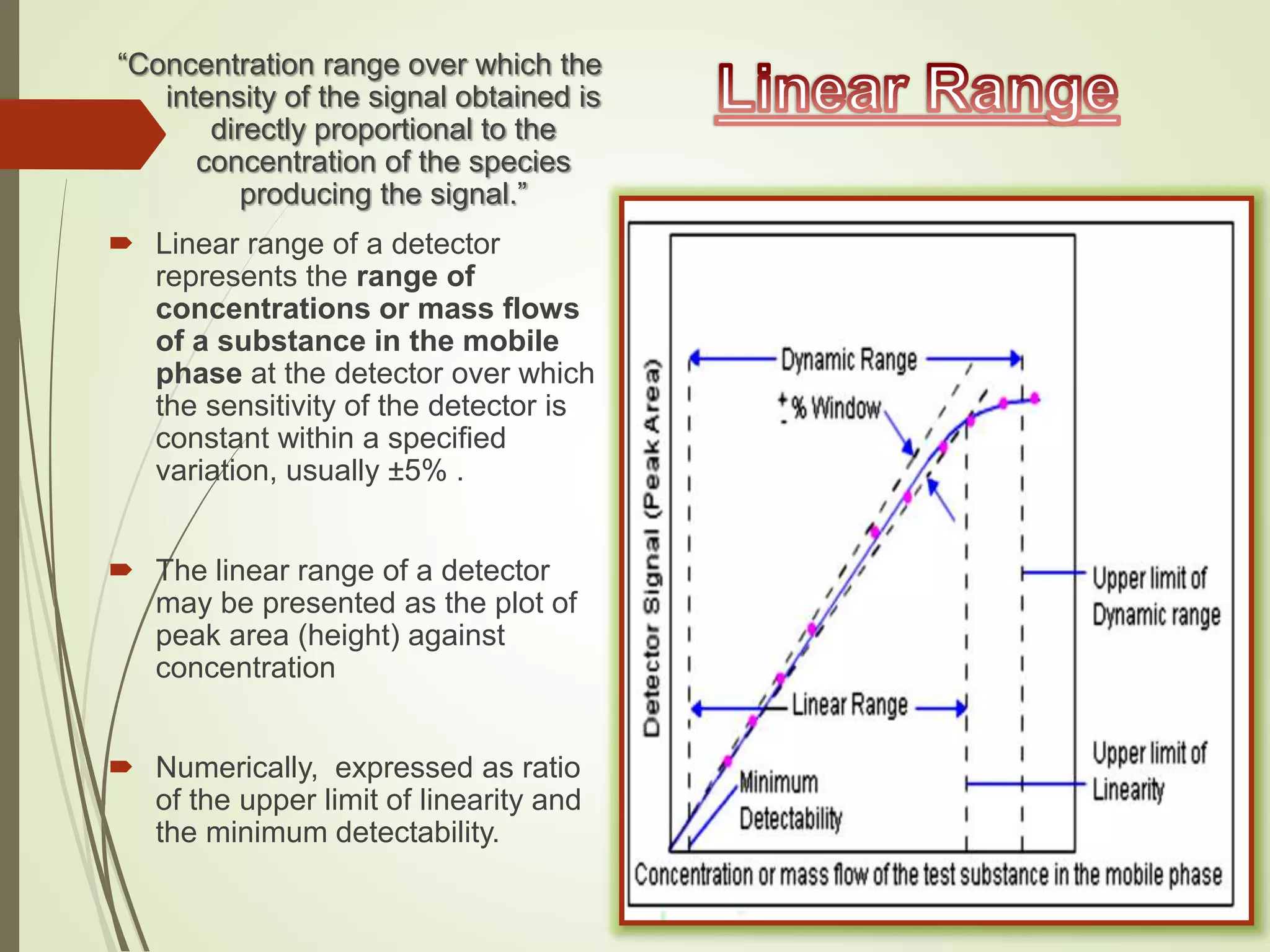






The document explains the qualitative and quantitative analysis techniques in High-Performance Liquid Chromatography (HPLC) and Gas Chromatography (GC) for determining the chemical constituents of substances. It discusses methods for peak identification using detectors and standards, the importance of calibration curves for quantification, and outlines validation parameters necessary for accurate analysis. Additionally, it emphasizes the role of HPLC in pharmaceutical development and the significance of method selection and detector functionality in the analytical process.



































































