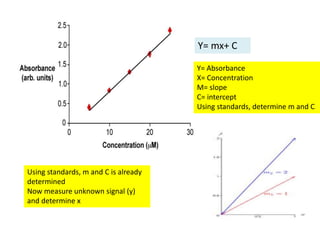
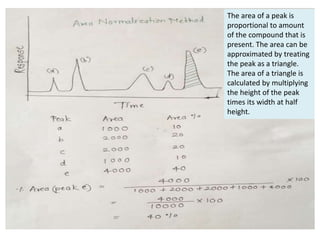
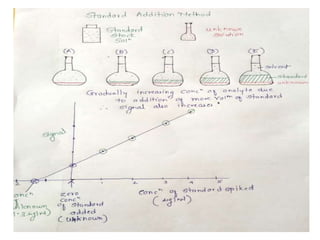
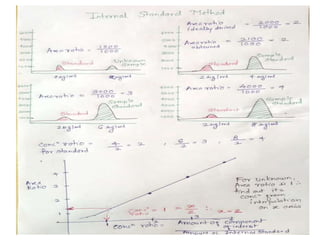
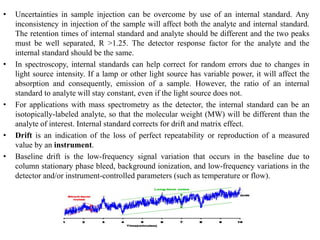
This document discusses various quantitative techniques used in chromatography, including external standard, internal standard, standard addition, and area normalization methods. The external standard method involves preparing calibration standards of known concentrations and constructing a calibration curve. The internal standard method uses an internal reference compound added to both samples and standards. The standard addition method determines an unknown concentration by adding increasing amounts of standard to the sample. Area normalization relies on the detector response being proportional to amount without reference standards. Multiple-point standardization and internal standards help account for matrix effects and instrument drift.