





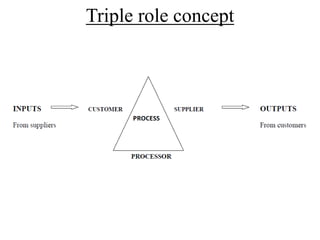















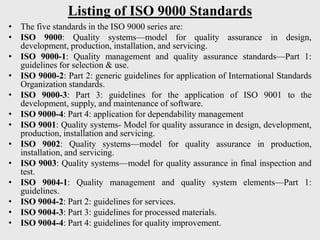













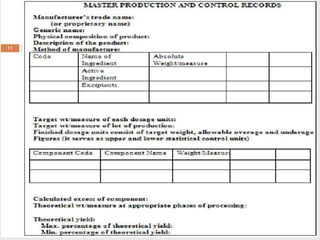


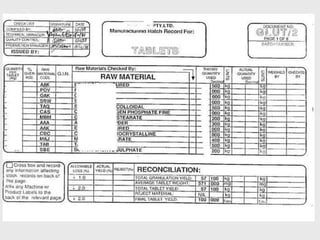















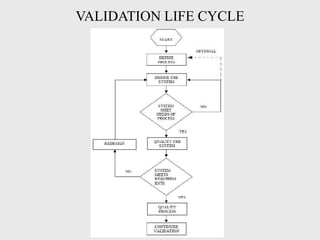




The document discusses various concepts related to quality including definitions of quality, quality management, quality control, quality assurance, ISO standards, total quality management, and documentation requirements. It provides definitions for quality as fitness for use, conformance to specifications, and meeting customer expectations. Quality management involves building quality into products through controls and preventing deficiencies. Quality control tests and inspects materials and products, while quality assurance reviews quality systems and procedures. Documentation is essential for defining and controlling quality systems.










































































