Downloaded 77 times






















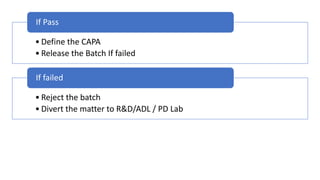






The document provides an in-depth overview of 'Out of Specifications' (OOS) results in regulatory practices, explaining the definition, causes, and investigation procedures as per MHRA and CDER guidelines. It details the phases of investigations, including laboratory and manufacturing inquiries, as well as protocols for re-sampling and re-analysis. Furthermore, it outlines the regulatory implications of OOS results, such as reporting requirements and restrictions on product handling and sales.



























![ICH [ Q ] Guidelines](https://cdn.slidesharecdn.com/ss_thumbnails/ichabhishek-210812054107-thumbnail.jpg?width=640&height=640&fit=bounds)







































