Downloaded 259 times










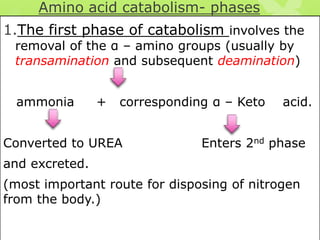
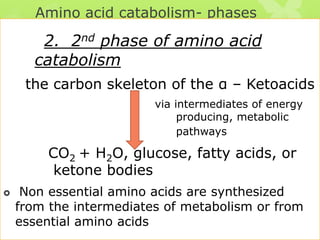

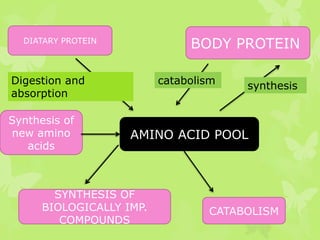
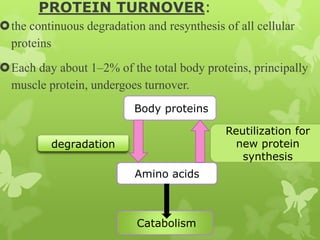

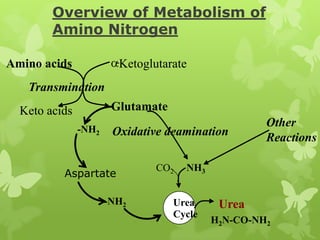

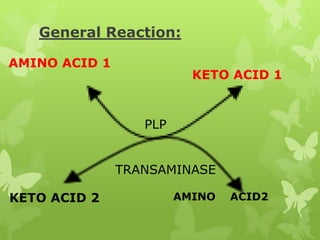

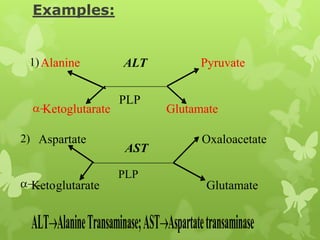












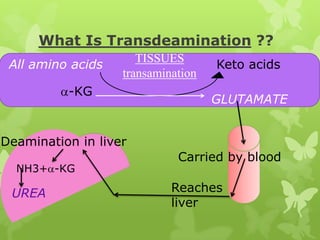





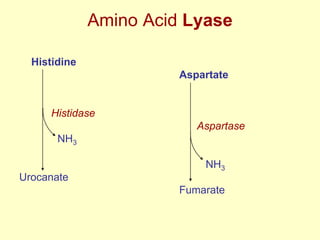
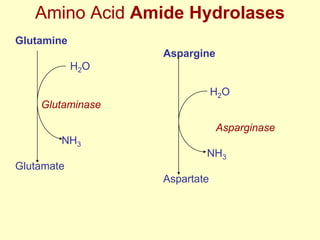



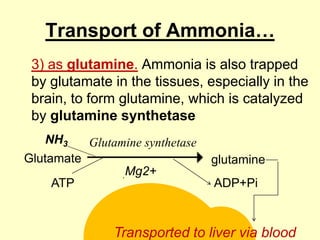
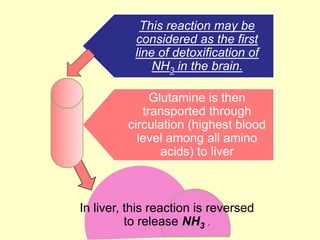
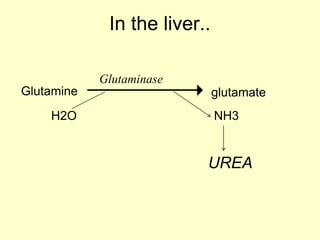
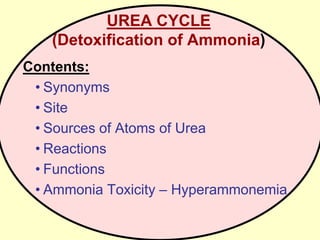


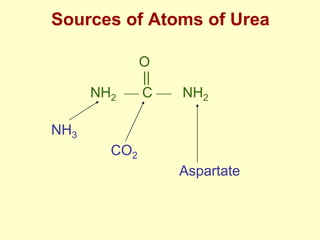

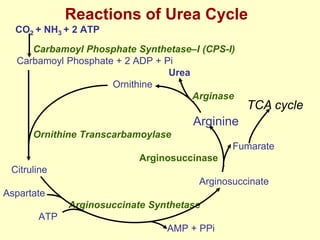








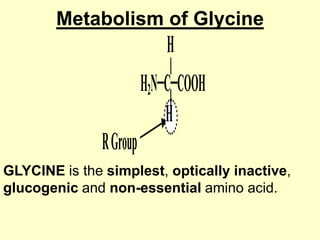



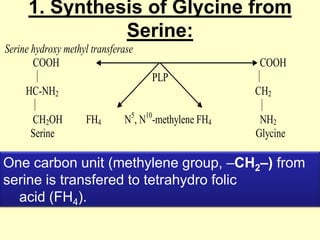
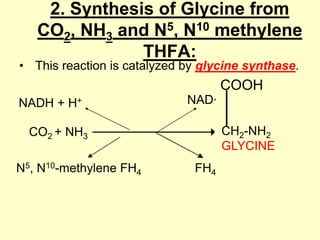
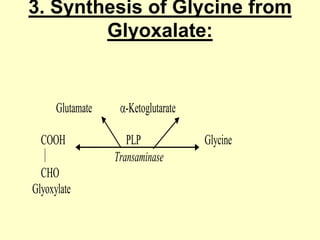
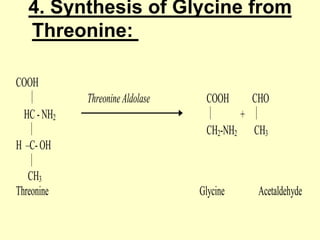

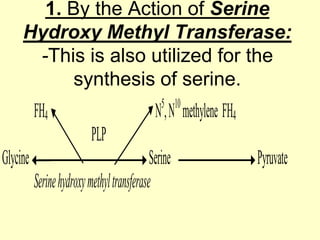
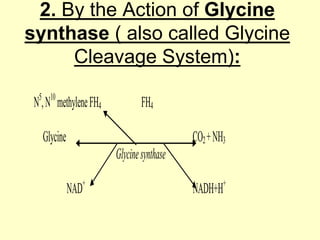
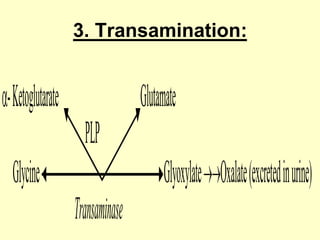

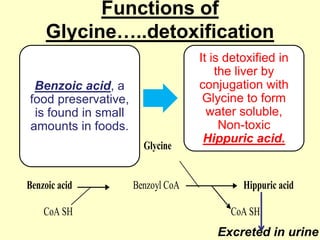



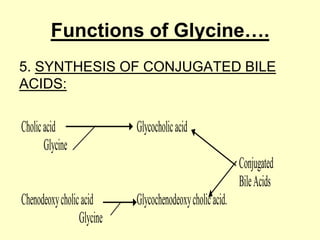

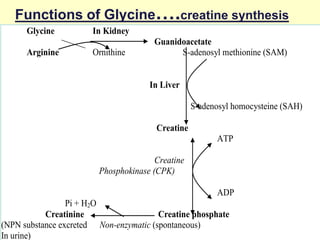

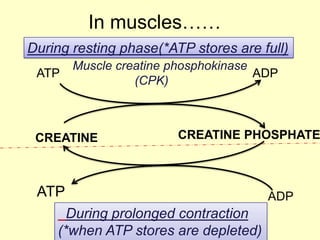

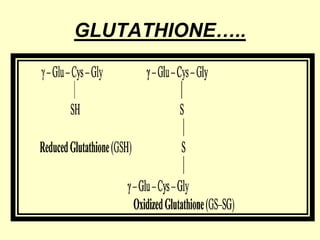
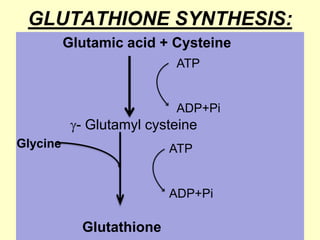




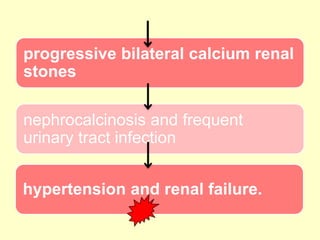
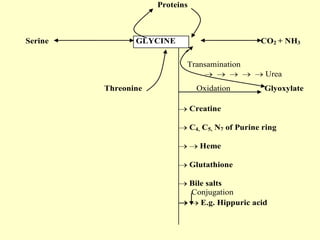




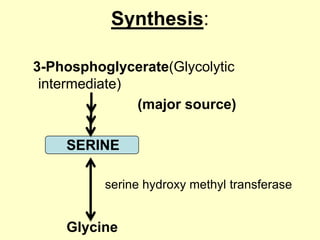




The document outlines protein and amino acid metabolism, covering essential processes such as the breakdown of proteins, amino acid catabolism phases, and the urea cycle which detoxifies ammonia. It highlights the biological importance of amino acids in protein synthesis and their metabolic pathways, including transamination and deamination. Clinical implications of amino acid metabolism disorders, including hyperammonemia and genetic defects, are also discussed.



















































































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)












