





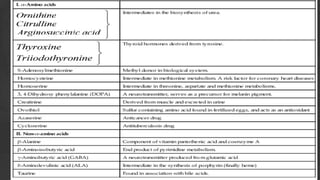
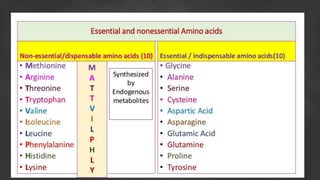
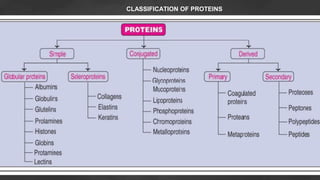

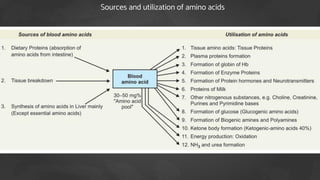


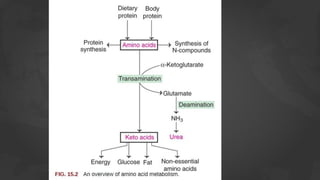
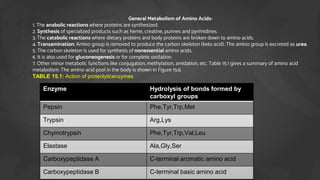
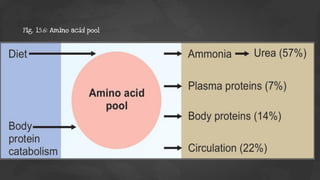

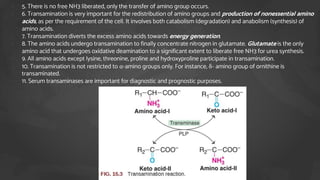











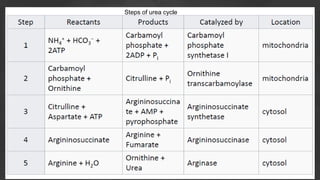



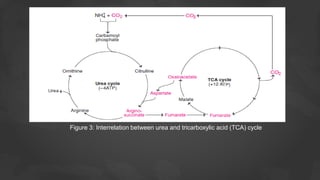



1. The document discusses amino acid and protein metabolism. It covers topics like the amino acid pool, transamination, deamination, the metabolism of ammonia, and the urea cycle. 2. Transamination is the process where the amino group is transferred from one amino acid to a keto acid, catalyzed by transaminase enzymes. Deamination results in the liberation of ammonia for urea synthesis and the conversion of the amino acid carbon skeleton into a keto acid. 3. Glutamate plays a central role as it can accept amino groups via transamination and also undergo oxidative deamination to release ammonia via glutamate dehydrogenase. This links amino acid and urea metabolism to the TCA












































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)
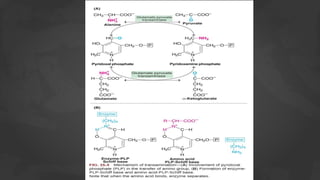
![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)









