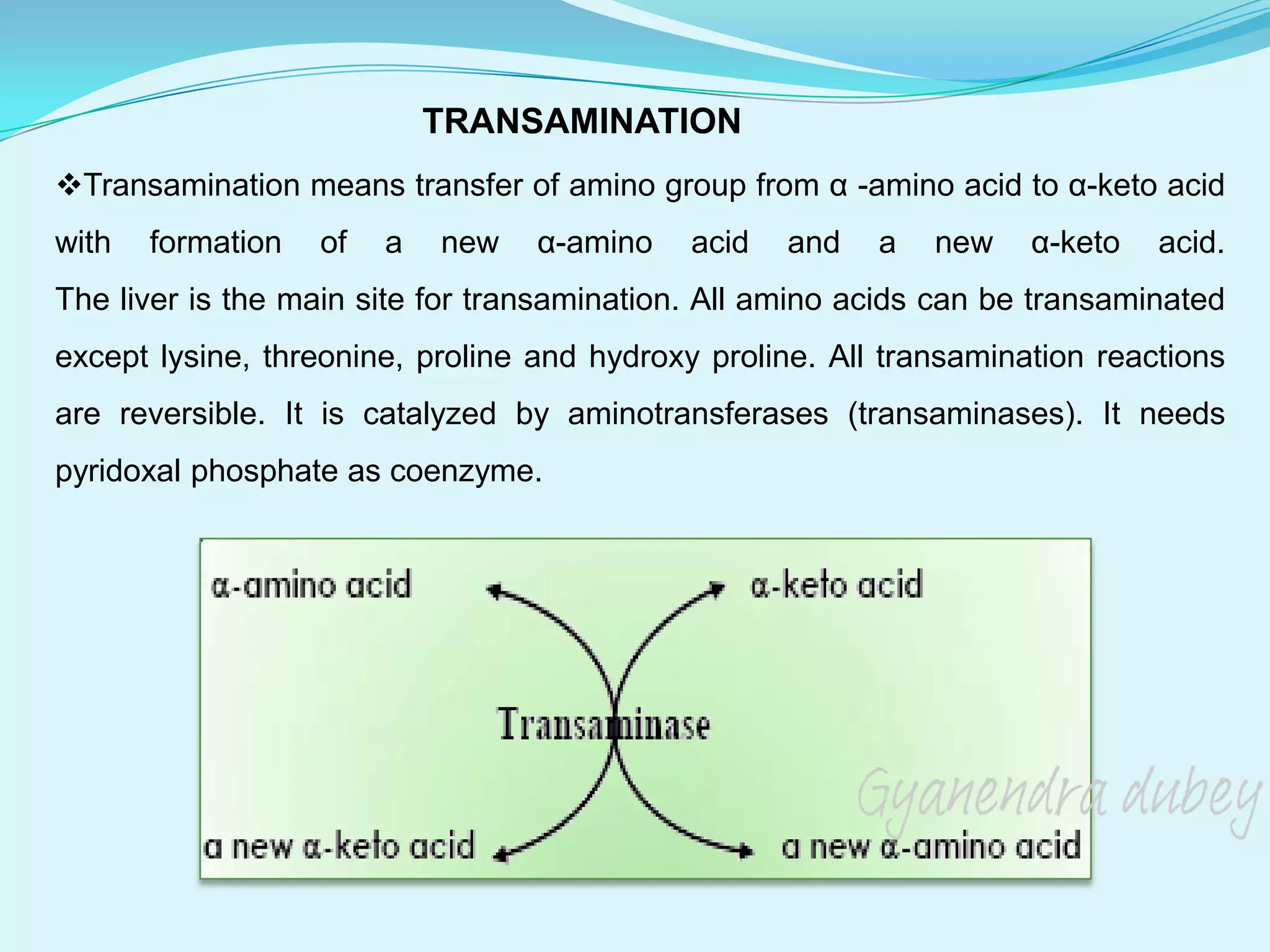
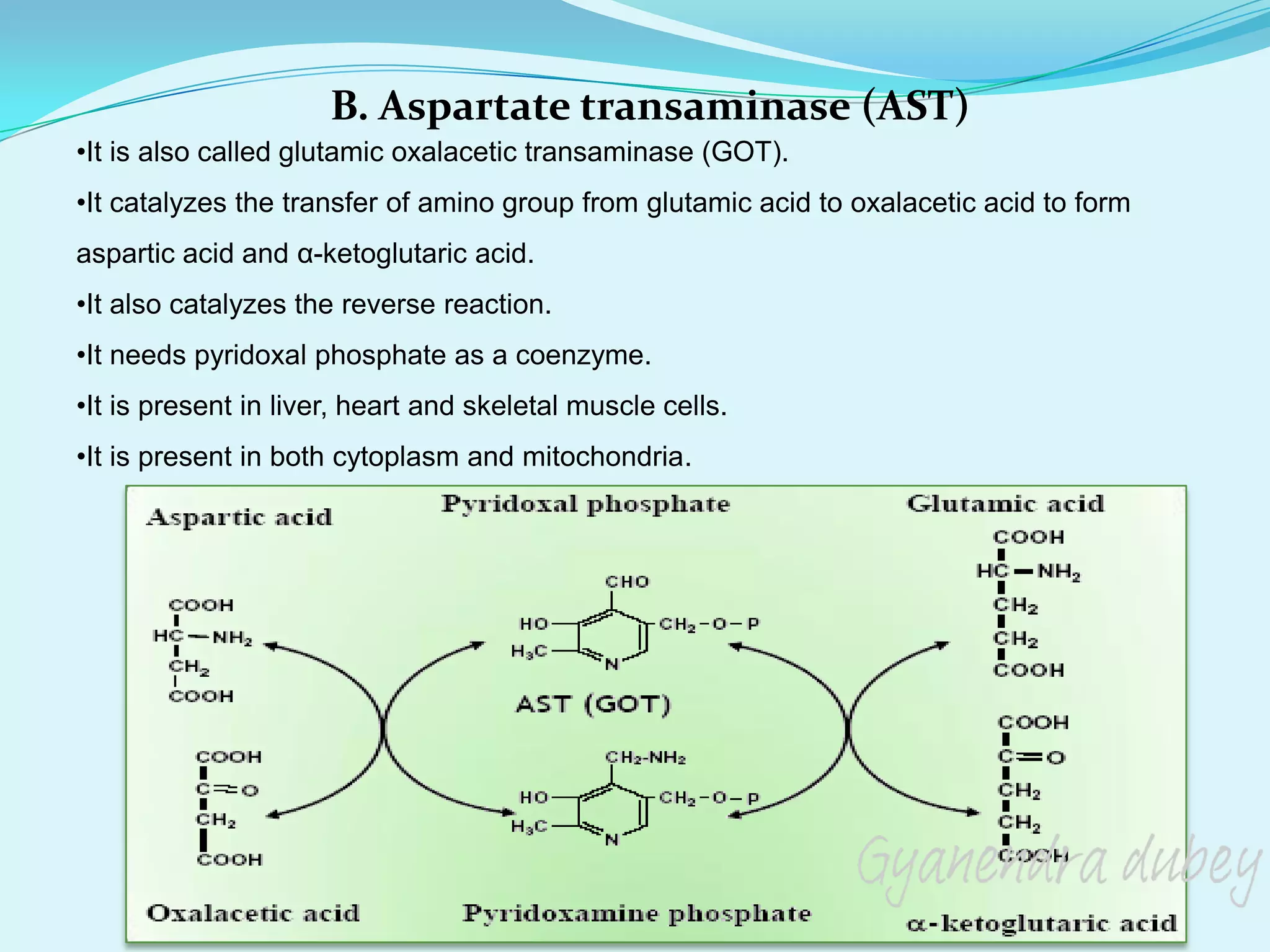
The document summarizes amino acid metabolism. It discusses how proteins are broken down into individual amino acids, which then undergo various reactions like transamination and deamination. Transamination involves transferring the amino group from an amino acid to a keto acid. Deamination removes the amino group as ammonia. The carbon skeleton is used for energy production, glucose synthesis, or fat/ketone body formation. Ammonia is converted to urea in the urea cycle in the liver to allow excretion. Deficiencies in urea cycle enzymes can cause metabolic disorders.