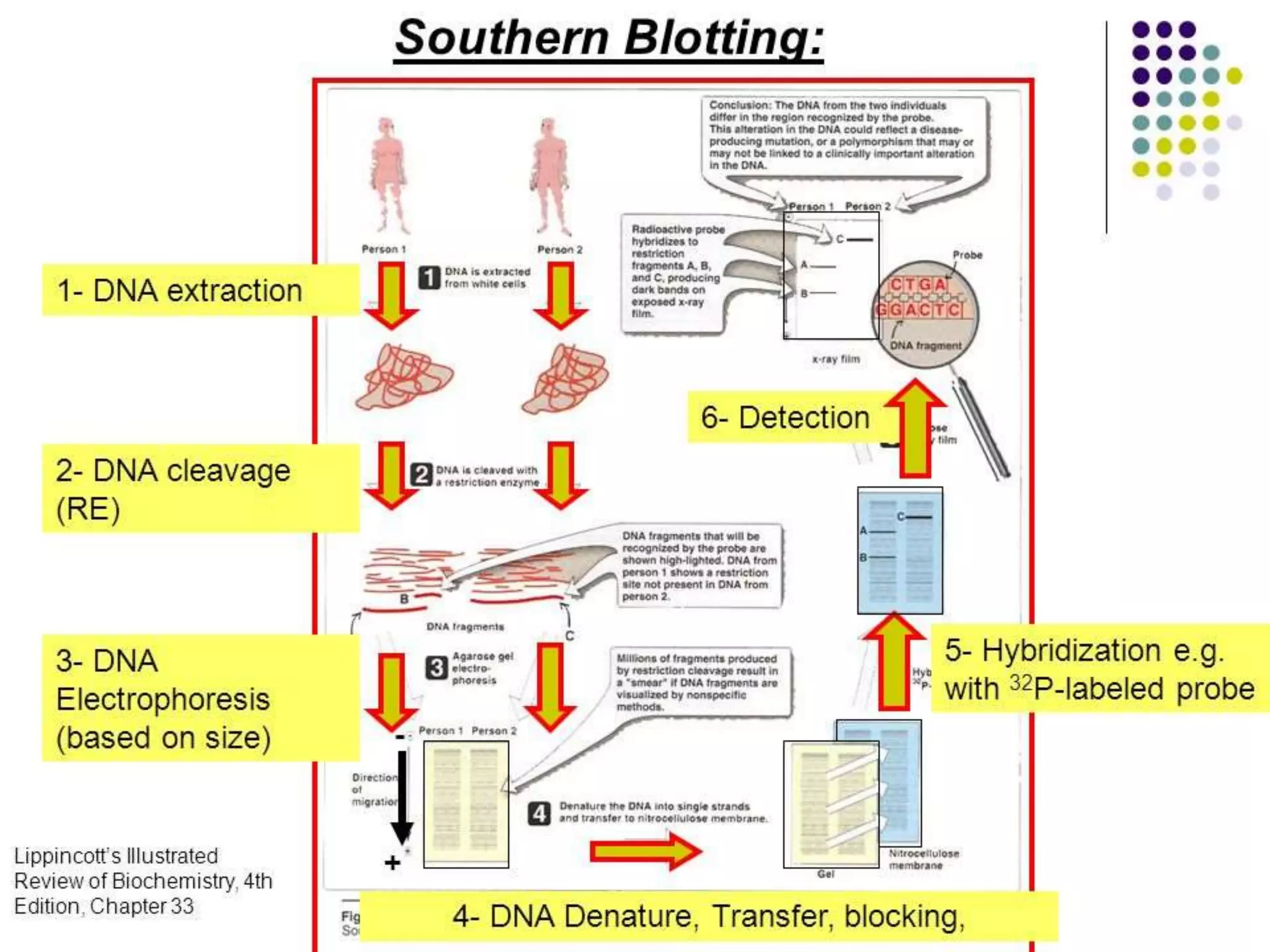
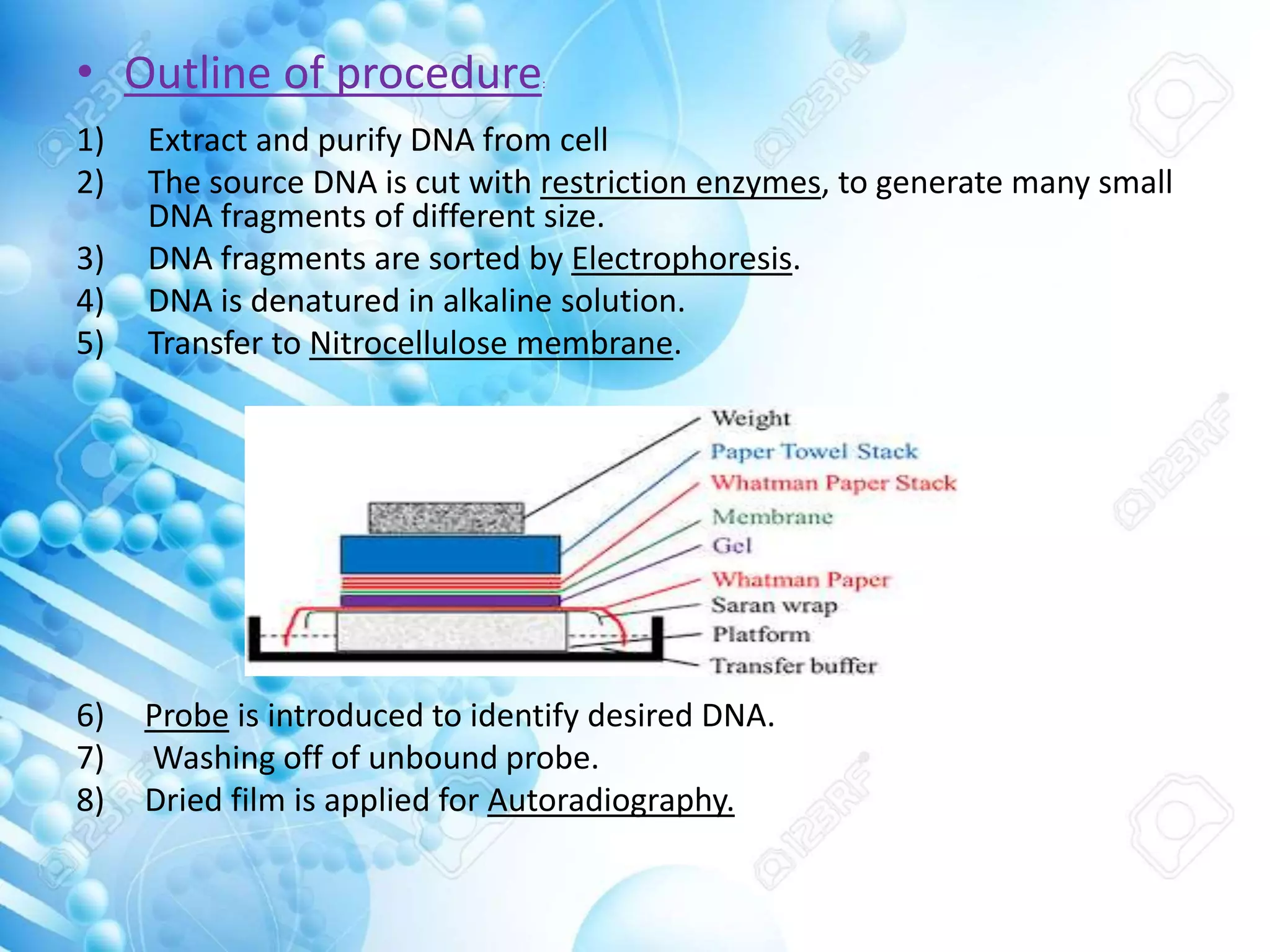
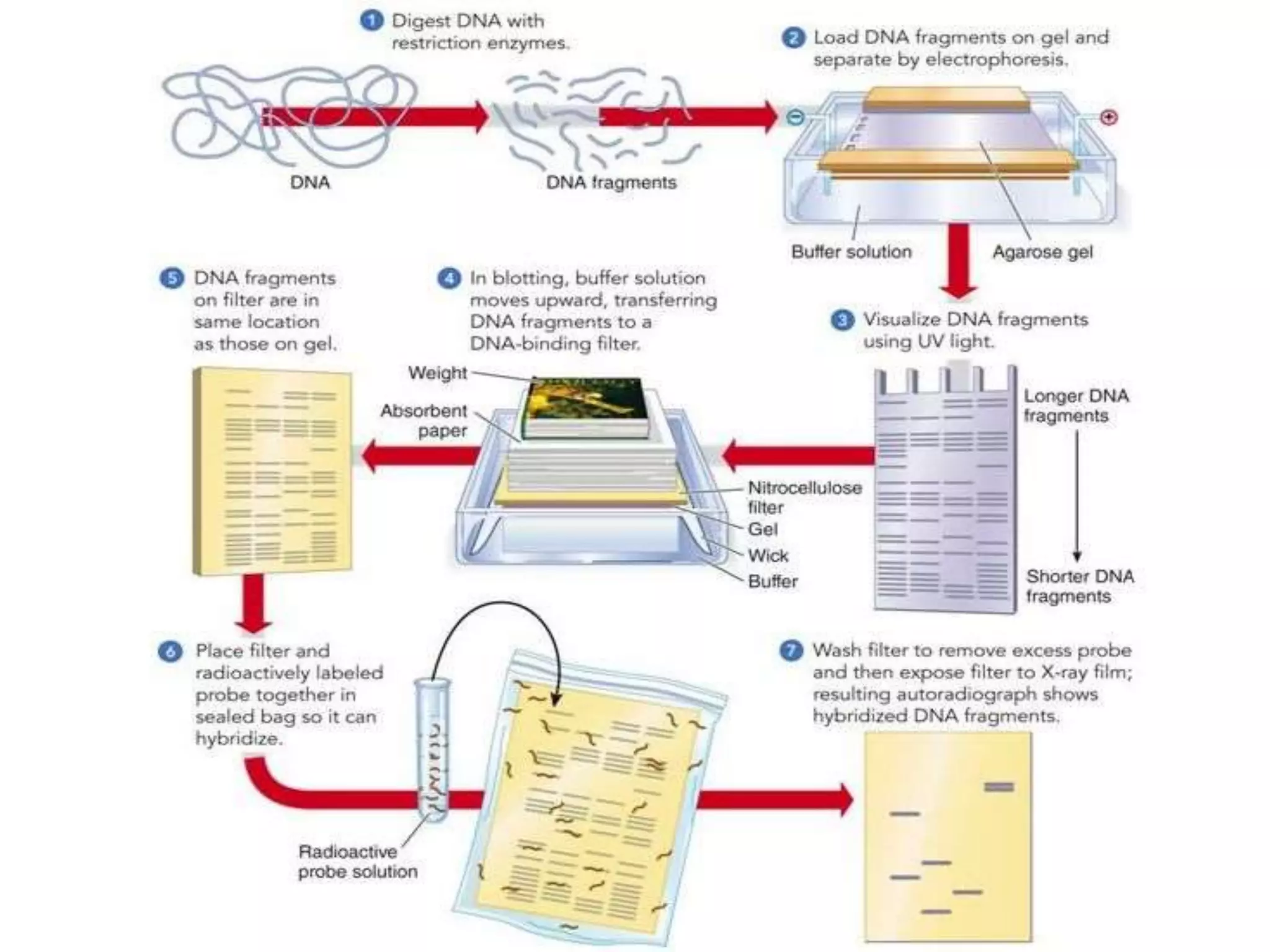
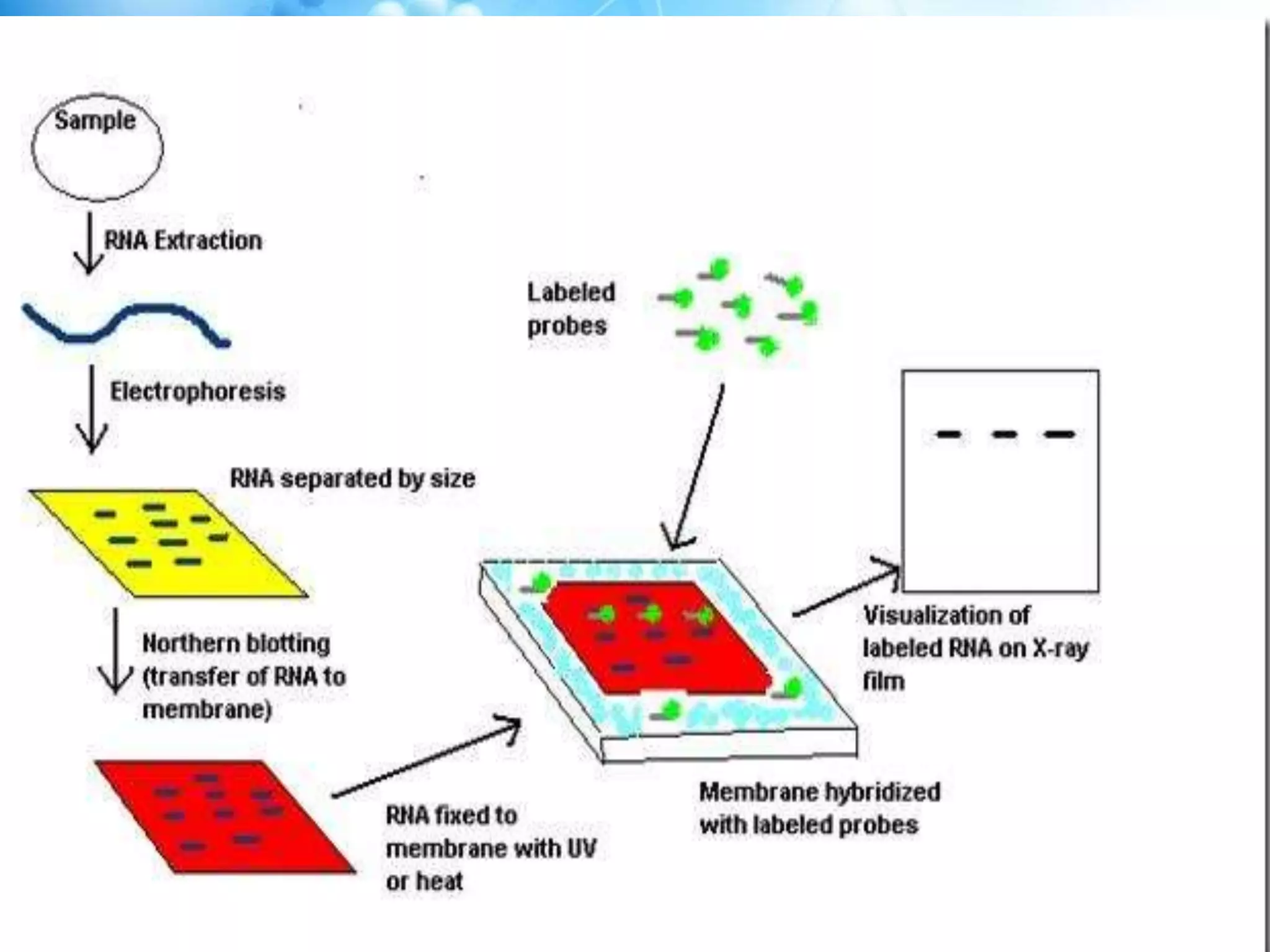
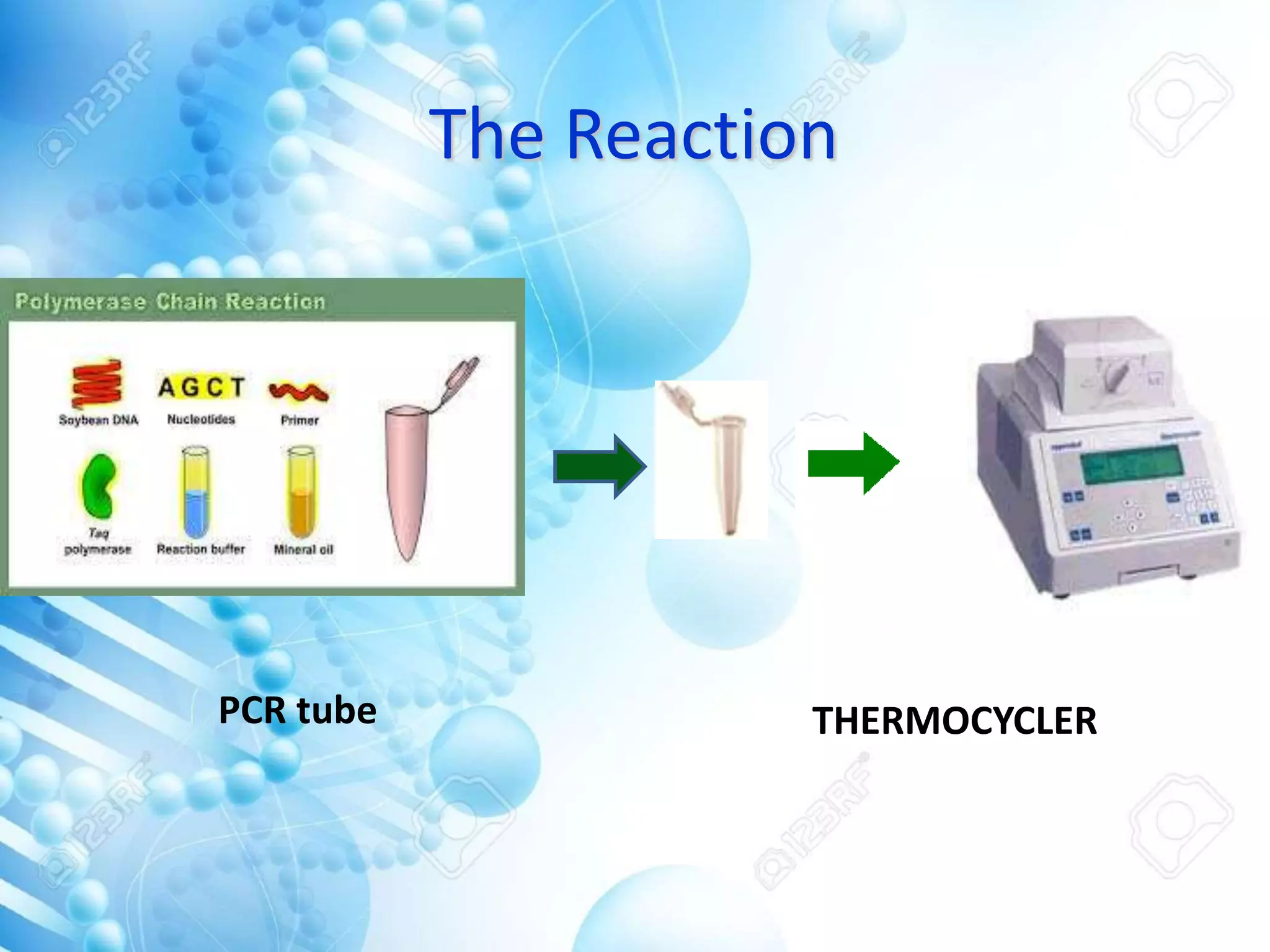
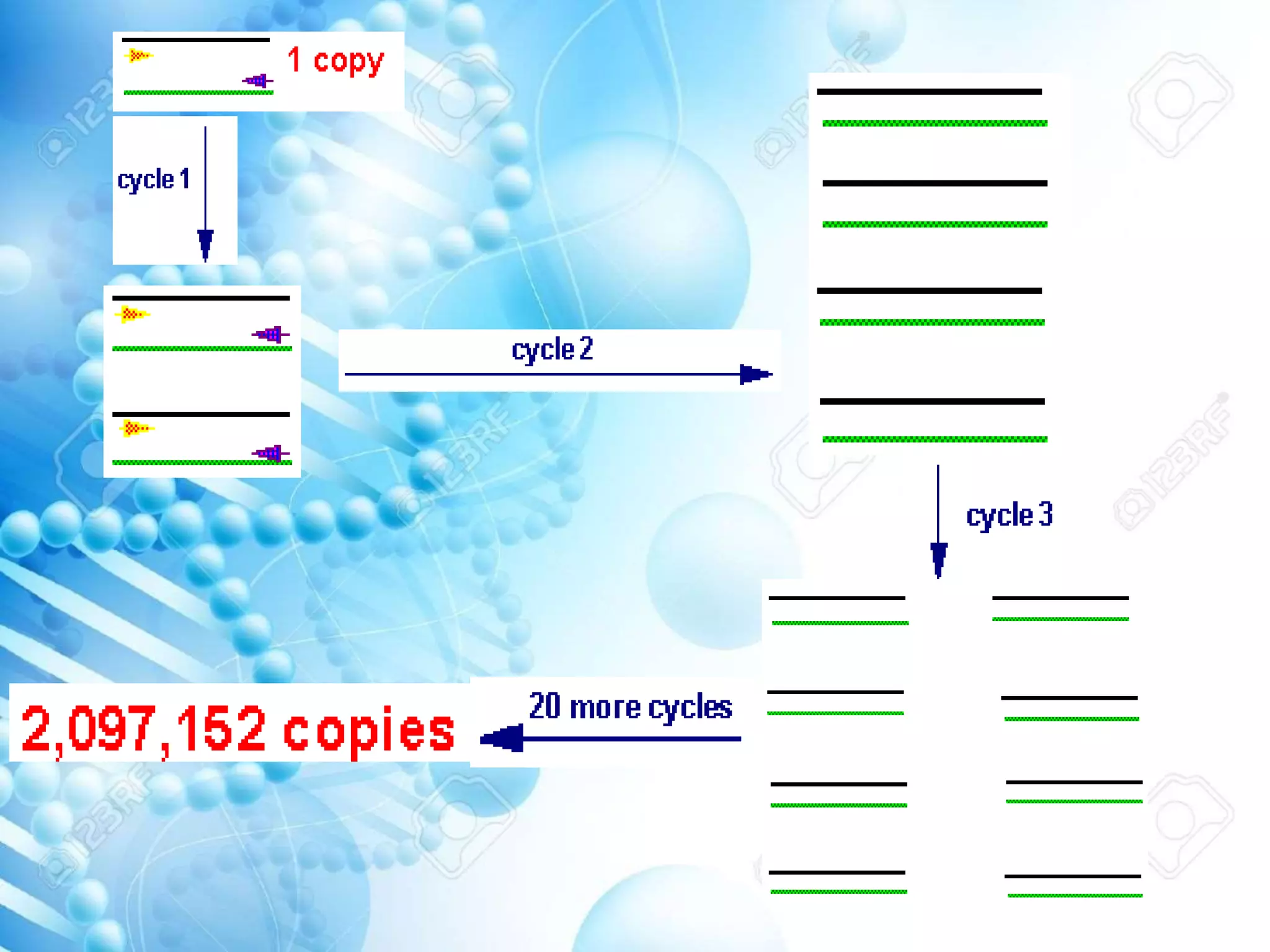
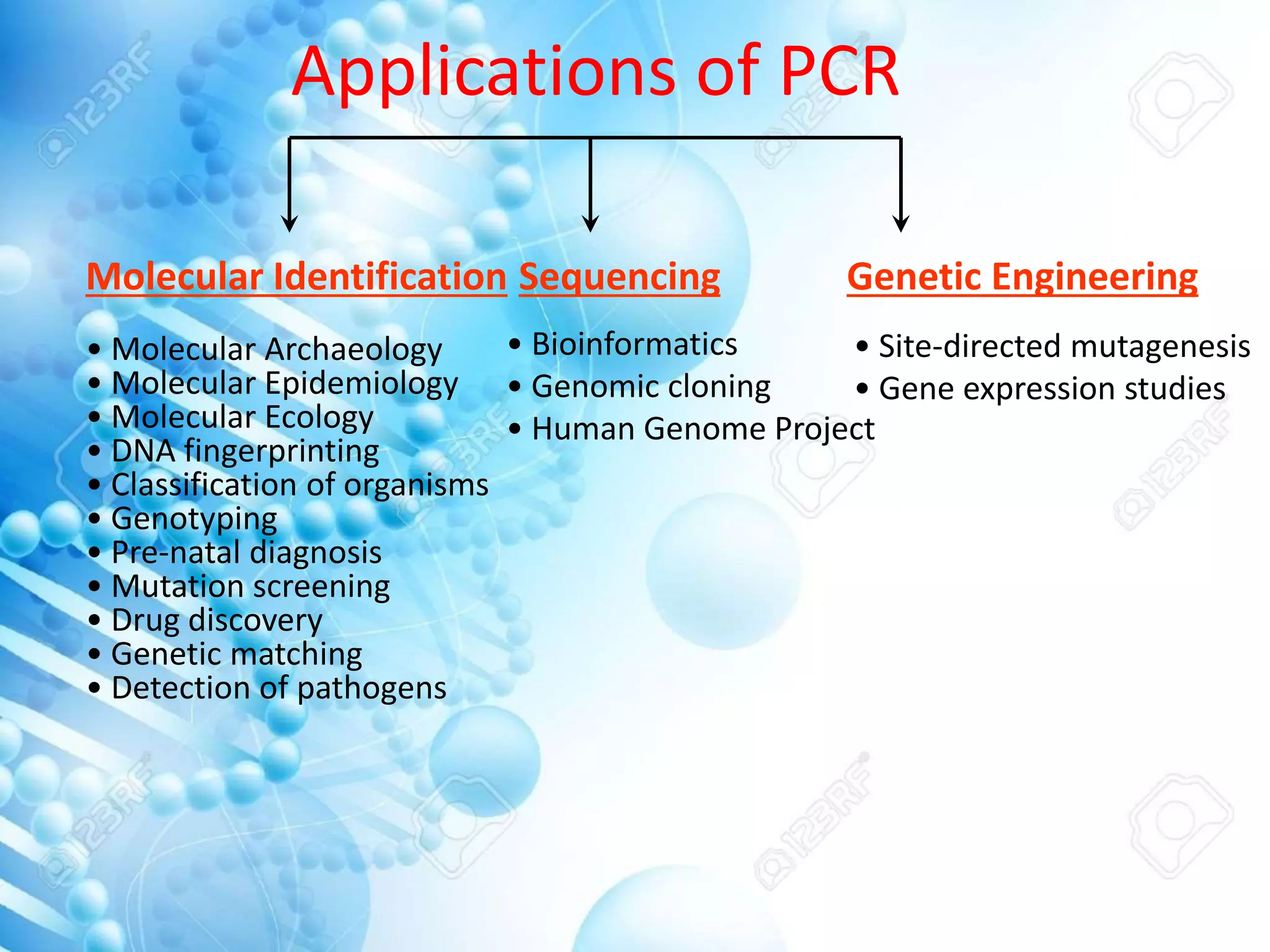
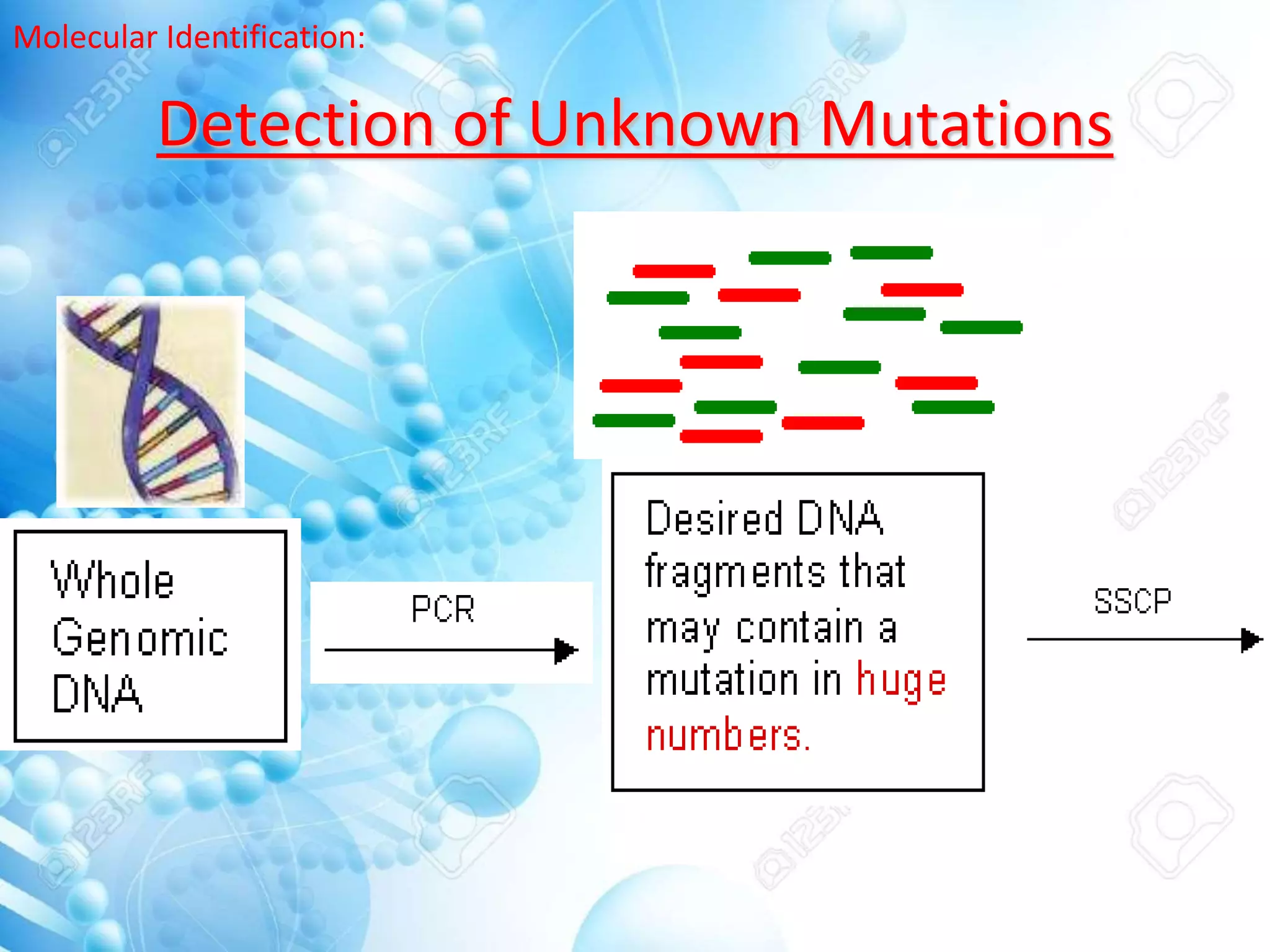
The document discusses various molecular techniques including blotting, probes, and polymerase chain reaction (PCR). It describes Southern blotting for detecting DNA, Northern blotting for RNA, and Western blotting for proteins. It explains how probes are used to identify specific DNA or RNA sequences in Southern and Northern blotting. The key steps of PCR are outlined, including denaturation, annealing of primers, and extension of DNA copies. Applications of these techniques include gene discovery, mutation detection, forensics, diagnosis of genetic disorders, and more. PCR has revolutionized research and diagnostics due to its speed, sensitivity, and specificity.





















































































![Histological development in enzymology [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/histologicaldevelopmentinenzymologyautosaved-170828131631-thumbnail.jpg?width=640&height=640&fit=bounds)



















