Downloaded 501 times




![Permanganate titration
KMnO4 Powerful oxidant that the most widely used.
Eq. Wt.(=M/5): In strongly acidic solutions (1M H2SO4 or HCl, pH ≤ 1)
MnO4– + 8H+ + 5e- = Mn2 + + 4H2 O
Eo = 1.51 V
violet color
colorless manganous
KMnO4 is a self-indicator.
In feebly acidic, neutral, or alkaline solutions (E=M/3)
MnO4– + 4H+ + 3e- = MnO2 (s) + 2H2 O
Eo = 0.59 V
brown manganese dioxide solid
In very strongly alkaline solution (2M NaOH or Ba (OH)2) (E=M/1)
MnO4– + e- = MnO42 –
Eo = 0.56 V
green manganate
III
E=M/4 (in HF or NH4HF2 Medium)
MnO4– + 4e- + 6F-+ 8H+ = [MnF6] 3 – + 4H2O
Trivalent Fluoro magnate anion](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-4-320.jpg)
![Estimation of Fe+2
In the analysis of iron ores, (solution is frequently effected in
conc. HCl); the Fe+3 is reduced and the Fe+2 is then determined in
the resultant solution.
If Cl- is present, to prevent its oxidation in acidic medium (1-2
N) by MnO4- about 25 mL of Zimmermann and Reinhardt's
solution (preventive solution) has to be used.
It is prepared by dissolving 50 g of crystallised (MnSO4,4H2O) in
250 mL water, adding a cooled mixture of 100 mL conc.H2SO4
and 300 mL water, followed by 100 mL H3PO4. The manganese
(II) sulphate (presence of Mn+2) lowers the oxidation potential of
the MnO4- - Mn(II) couple (-1.20V) and thereby makes it a weaker
oxidising agent; the tendency of the permanganate ion to oxidise
chloride ion is thus reduced.( Eo of Cl-/Cl2 is much higher)
[
] ]
0.0591
MnO 4 [ H
E = -1.52 log
[ Mn + 2 ]
5
−
+ 8
positive
See Vogels book](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-5-320.jpg)



![Standardization of KMnO4 solution
Standardization by titration of sodium oxalate Na2C2O4.2H20 (primary standard)
(Fowler and Bright) :
C2O42- = 2CO2 + 2 e-
E° red = +0.77V
2KMnO4 +5 Na2(COO)2 +8H2SO4 = 2MnSO4 +K2SO4 +5Na2SO4 +10 CO2 + 8H2O
The reaction between oxalic acid and potassium permanganate can be represented
as:
2KMnO4 + 5 H2C2O4 +3H2SO4 = 2MnSO4 +K2SO4 +10 CO2+ 8H2O
In ionic form the reaction can be represented as:
2MnO4- + 5 C2O4 2- +
16H+
= 2Mn2+ + 10 CO2 + 8H2O
This titration is carried out in warm conditions (60 oC). The reaction at room
temperature is slow because of the equilibrium nature of this reaction. CO2 is highly
soluble in water and thus heating removes all dissolved CO2 out of the solution
driving the reaction in forward direction.
Also at low temperature, the reduction of permanganate may not be
complete producing Mn(III) (in the form [Mn(C2O4)3]3-). The formation of this
species introduce errors in titrations as no. of electrons utilized here are different as
compared to production of Mn2+.](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-9-320.jpg)



![[
]
0.0591
Cr2 O 7 [ H
E =E log
[Cr +3 ]
6
0
−2
(10 )(10
0.0591
E = −1.33 log
-2 2
6
[10 ]
−3
0.0591
E = −1.33 log10 - 27
6
0.0591
E = −1.33 + 27 x
6
E = − .06V
1
]
+ 14
−2
)
14](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-13-320.jpg)
![E =E
[ Fe ]
- 0.0591log
[ Fe ]
+3
0
+2
0.003
E = -0.771 - 0.0591log
0.15
E = -0.671V
[
]
0.0591
MnO 4 [ H + ]
0
E=E log
[ Mn + 2 ]
5
−
8
( 0.02)( 1.00 )
0.0591
E = -1.51 log
( 0.005)
5
E = -1.52 V
8](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-14-320.jpg)







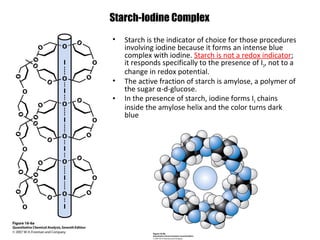
![Starch-Iodine complex
Starch solution(05~ 1%) is not redox indicator.
The active fraction of starch is amylose, a polymer of the sugar αD-glucose ( 1,4 bond).
The polymer exists as a coiled helix into which small molecules
can fit.
In the presence of starch and I–, iodine molecules form long
chains of I5– ions that occupy the center of the amylose helix.
••••[I I I I I]– ••••[I I I I I]– ••••
Visible absorption by the I5– chain bound within the helix gives
rise to the characteristic starch-iodine color.](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-23-320.jpg)


![Determination of Cu+2 :
2Cu+2 + 4I-
→ CuI + I2
Acetic acid buffer pH ~4.5 or better NH4HF2 buffer. In
presence of free mineral acid, at pH<3, dissolved O 2
liberate I2 from I- also.
The elments which interferes with the iodometric
determination are iron, arsenic and antimony, Trivalent
iron is reduced by iodide:
2Fe3+ + 2I- ⇋ 2Fe2+ + I2
but by addition of excess of fluoride, the iron(III) is
converted into the complex [FeF6]3-, which yields so small
a concn of Fe+3 ions that it has no oxidising action upon the](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-29-320.jpg)
![DETERMINATION OF THE AVAILABLE CHLORINE IN Bleaching powder
the hypochlorite solution or suspension is treated with an
excess of a solution of potassium iodide, and strongly
acidified with acetic acid:
Ca(OCl)+ KI +HAc → CaCl2 + I2 + H2O + KAc
The liberated iodine is titrated with standard sodium
thiosulphate solution.
Determination of hypochlorite in bleaches [CaCl(OCl)H2O]:
OCl– + 2I– + 2H+ → Cl– + I2 + H2O
(unmeasured excess KI)
I2 + 2 S2O3 2– → 2I– + S4O6 2–
Indicator: soluble starch (β-amylose)](https://image.slidesharecdn.com/permanganometryiodometryinanalyticaltechnique-140120005521-phpapp01/85/Permanganometry-iodometry-in-analytical-technique-P-K-MANI-30-320.jpg)



This document provides information about various redox titration methods including permanganometry, dichromatometry, iodometry, and iodimetry. It discusses the standard redox potentials and reaction equations for potassium permanganate, potassium dichromate, iodine, and other oxidizing agents. The document also describes procedures for standardizing and preparing standard solutions of these titrants. Specific applications discussed include the titration of iron(II), nitrites, and arsenite.

































































































