Downloaded 179 times


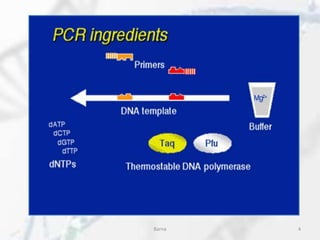
![Chemical components:
•DNA template that contains the DNA region (target) to be
amplified.
•Two primers that are complementary to the 3' (three prime) ends of
each of the sense and anti-sense strand of the DNA target.
•Taq polymerase or another DNA polymerase with a temperature
optimum at around 70 °C.
•Deoxynucleoside triphosphates (dNTPs) nucleotides containing
triphosphate groups), the building-blocks from which the DNA
polymerase synthesizes a new DNA strand.
•Buffer solution, providing a suitable chemical environment for
optimum activity and stability of the DNA polymerase.
•Divalent cations, magnesium or manganese ions; generally Mg2+ is
used, but Mn2+ can be utilized for PCR-mediated DNA
mutagenesis, as higher Mn2+ concentration increases the error rate
during DNA synthesis[7]
•Monovalent cation potassium ions.
Barna 3](https://image.slidesharecdn.com/pcr-150914050347-lva1-app6891/85/Pcr-3-320.jpg)






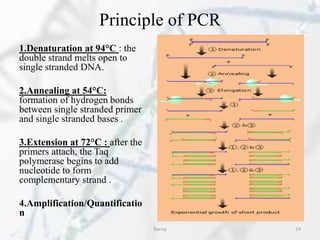
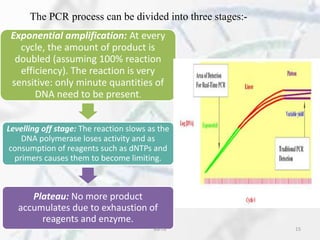

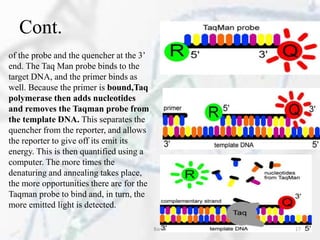

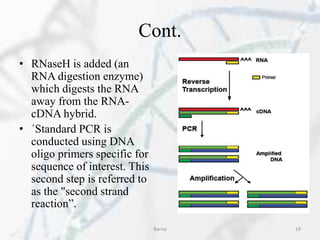

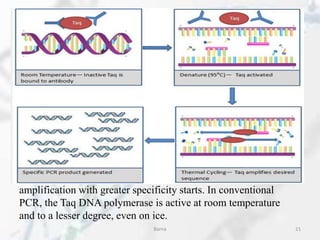
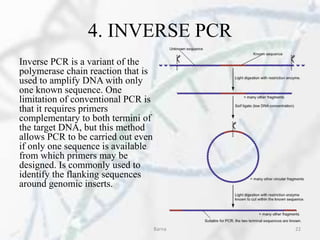
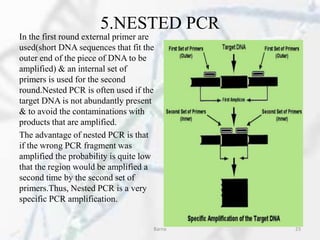


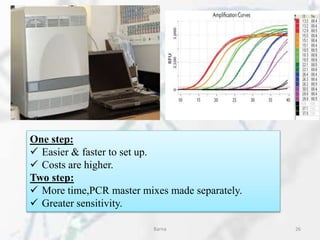






The polymerase chain reaction (PCR) is a technique used to amplify specific DNA fragments. It involves repeated cycles of heating and cooling of the DNA sample in the presence of DNA polymerase, primers, and nucleotides. During each cycle, the DNA strand is separated from its complement at a high temperature and two new strands are synthesized from the original copies at a lower temperature, thereby exponentially increasing the number of target DNA copies. Real-time PCR allows for quantification of the target DNA by detecting fluorescence at each cycle, while reverse transcription PCR is used to transcribe RNA into DNA.





























































































