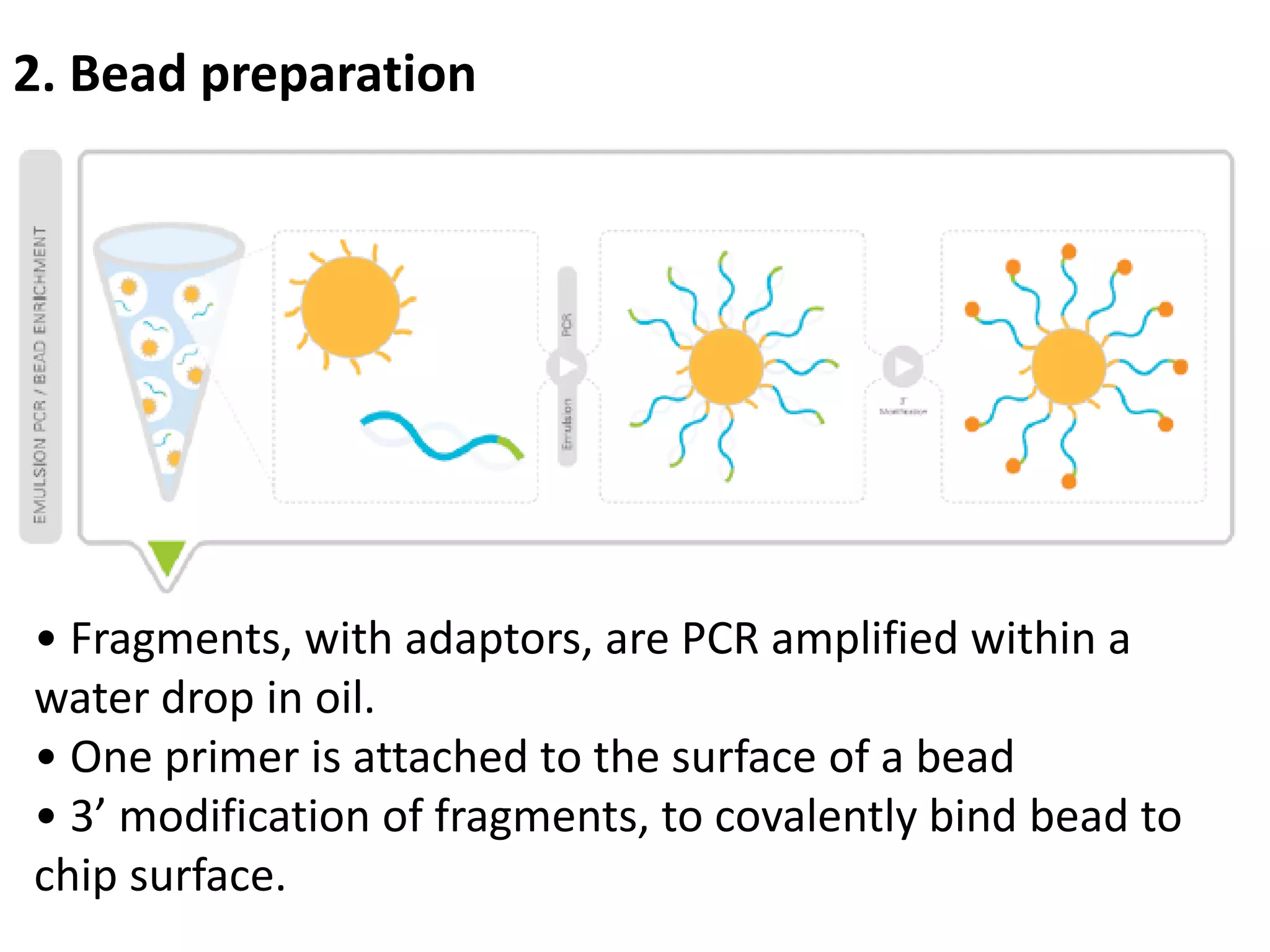
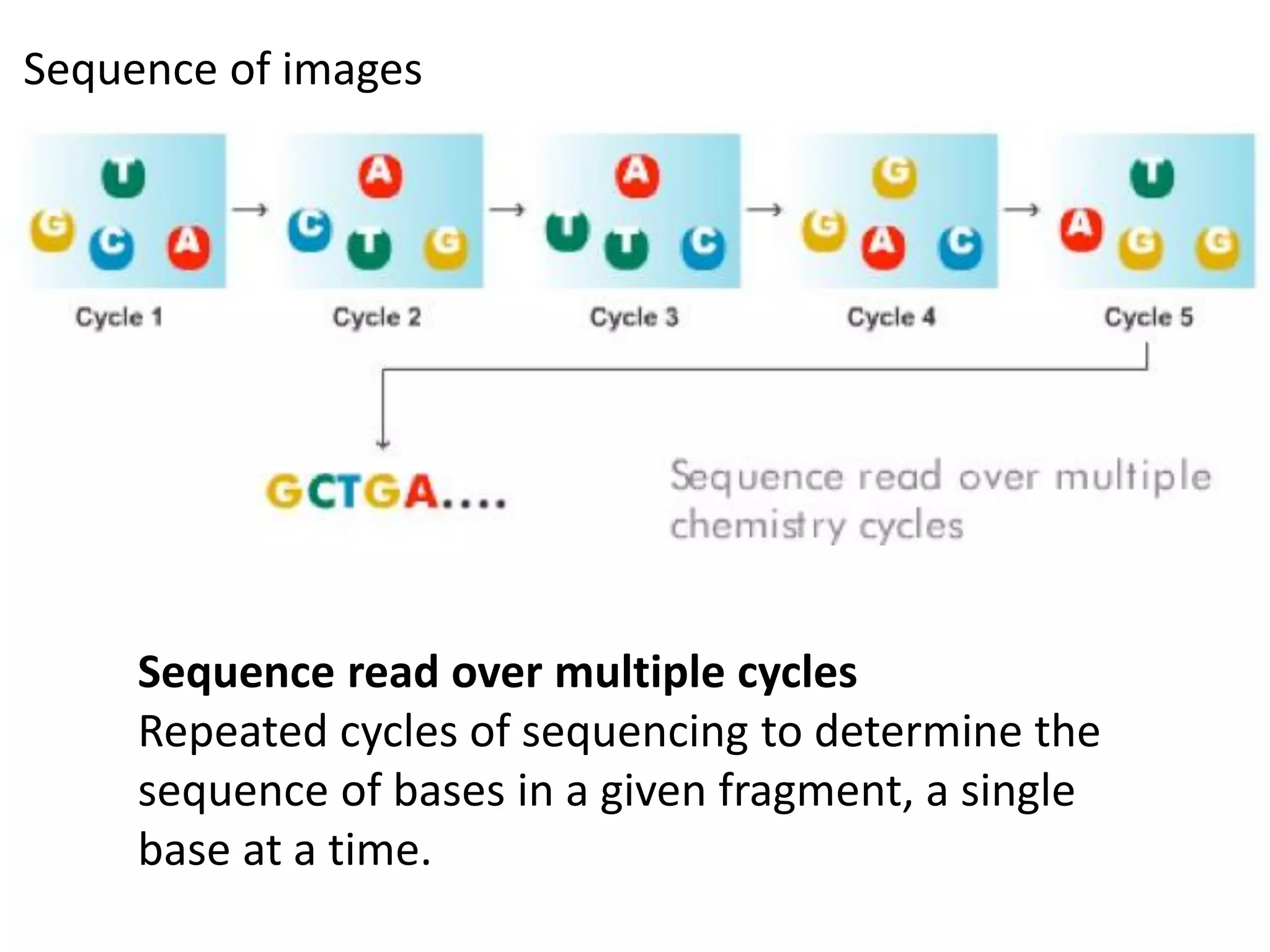
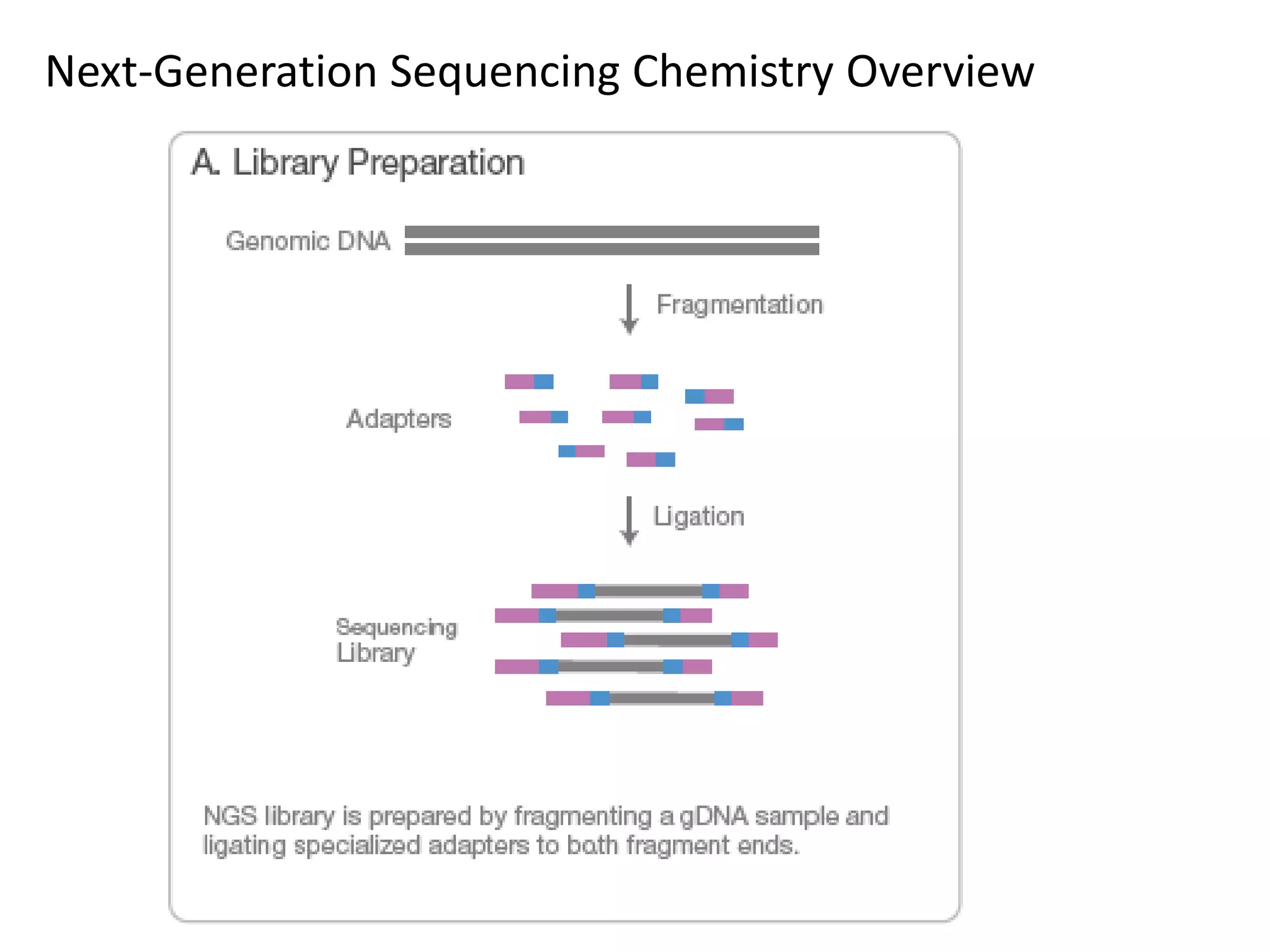
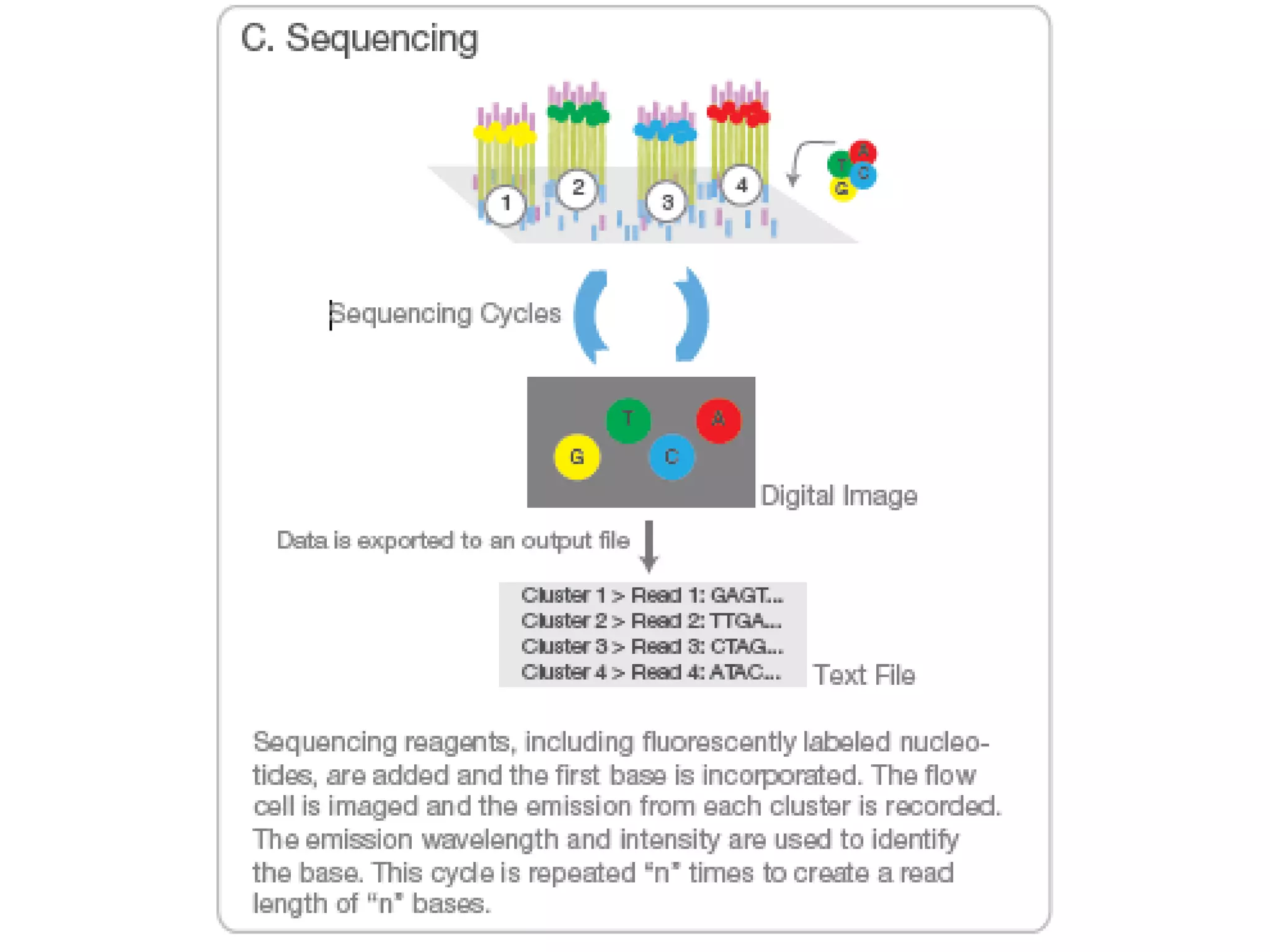
Next-generation sequencing (NGS) is a modern DNA sequencing technology that offers advantages over traditional Sanger sequencing in terms of speed, cost, sample size, and accuracy. NGS allows for massive parallel sequencing, drastically reducing the time and financial resources required for genomic studies while enabling high-throughput analysis of genetic material. Technologies such as Illumina sequencing and Ion Torrent have revolutionized the field, allowing for the sequencing of entire human genomes at unprecedented speeds and lower costs.