Download to read offline



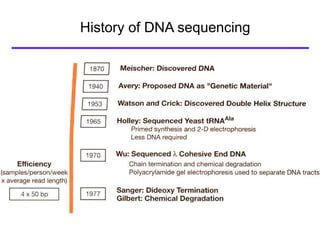
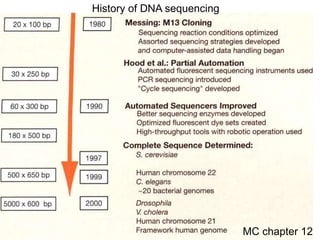




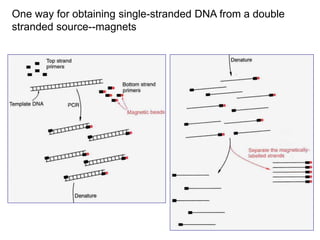

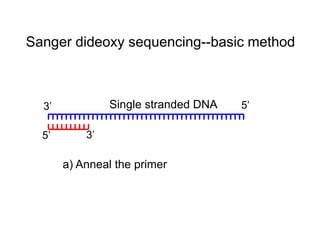
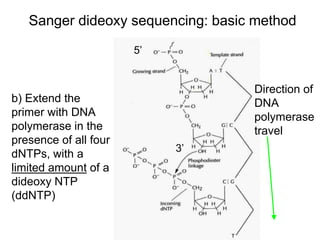

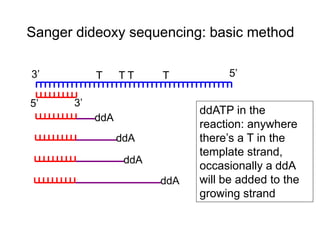
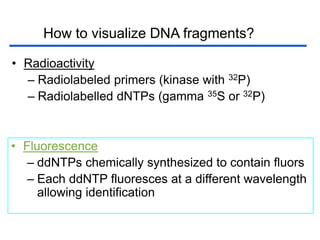

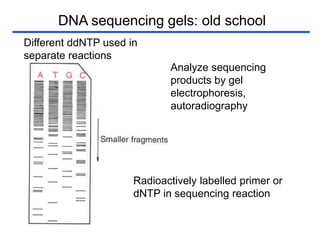
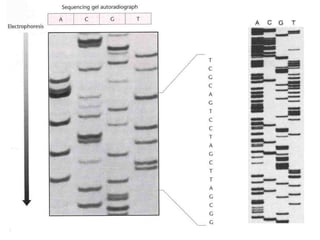
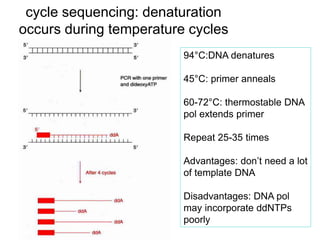

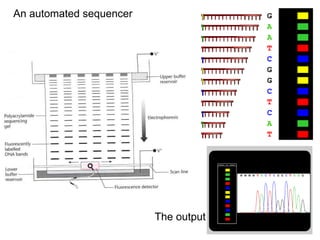

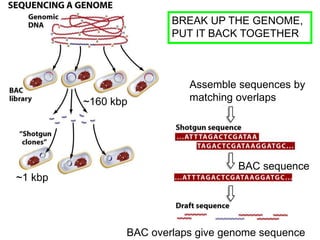





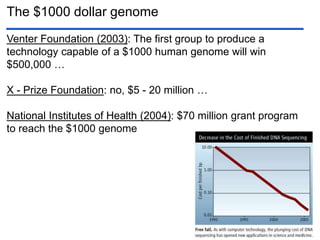


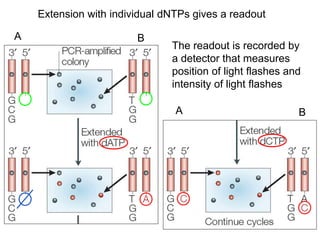
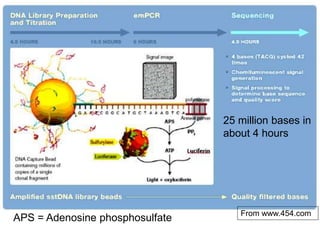
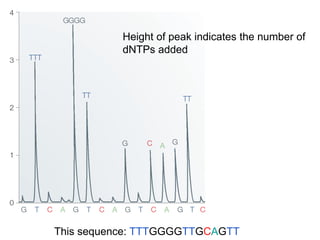



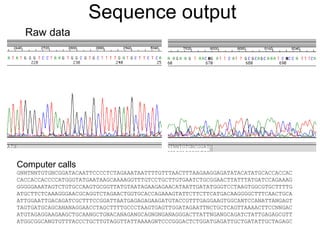
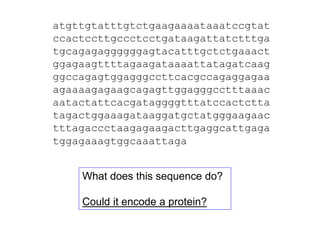
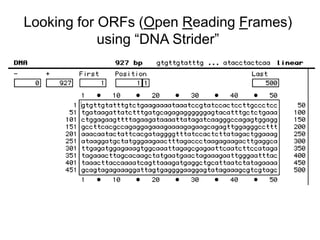
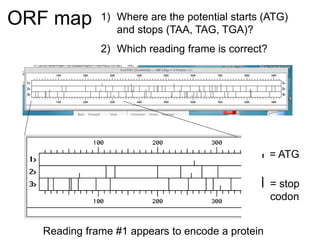






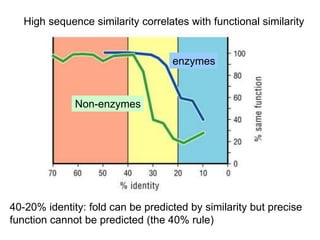




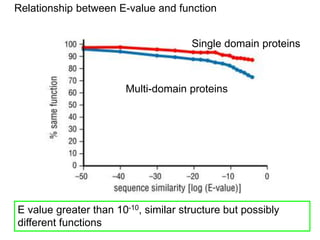
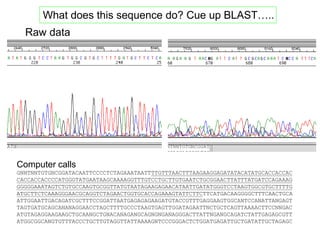









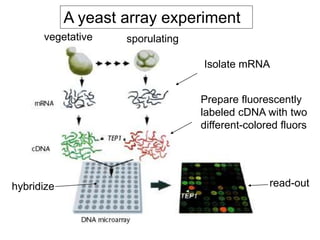
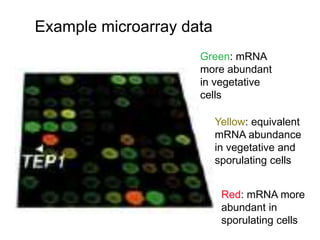

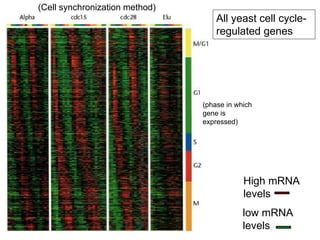

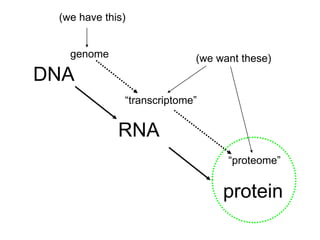



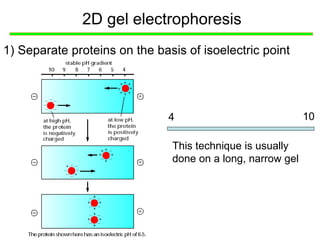
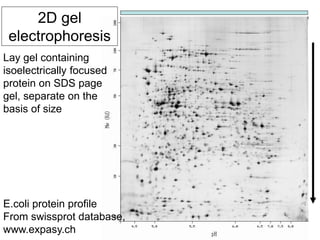
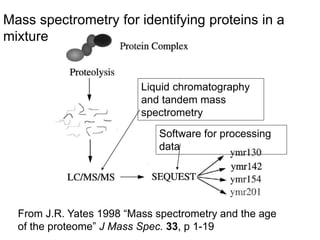


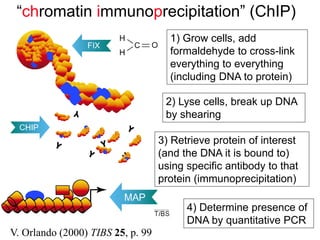
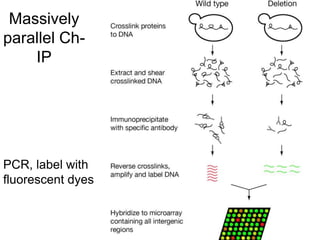
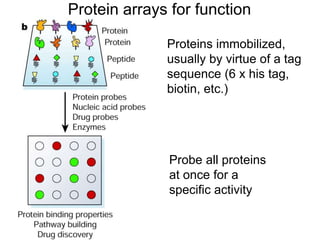

I. The document provides an overview of DNA sequencing methods, including a brief history and discussion of the Sanger dideoxy method, sequencing large pieces of DNA using shotgun sequencing, and progress towards achieving the "$1,000 genome". II. It describes the Sanger dideoxy chain termination method and how primers, templates, and reagents are used. Newer methods like pyrosequencing that can sequence many DNA molecules in parallel are also covered. III. The document discusses how sequenced DNA can be assembled and annotated, and tools for identifying genes and predicting functions like BLAST searches of databases. Reducing the cost of genome sequencing enables more widespread applications.


























































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)