Downloaded 319 times










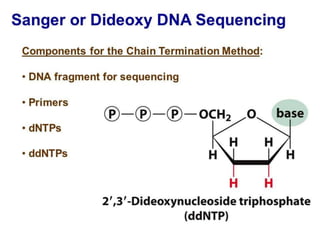
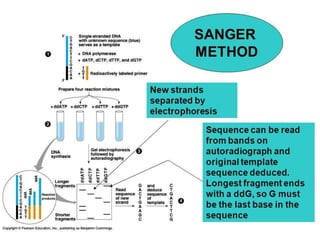
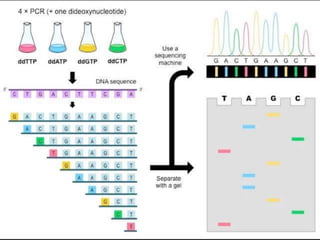

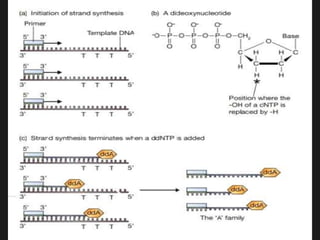

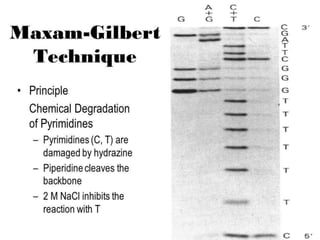











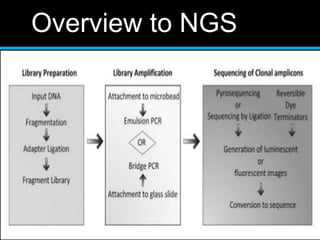


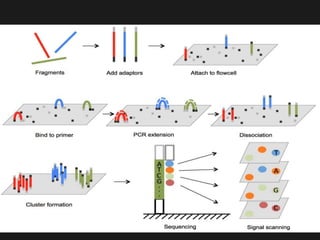

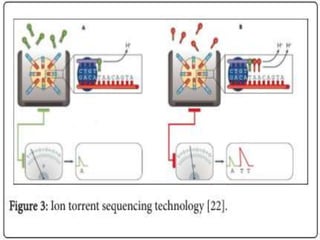
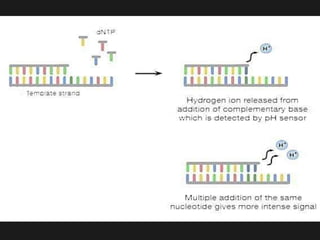

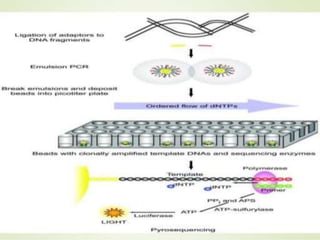



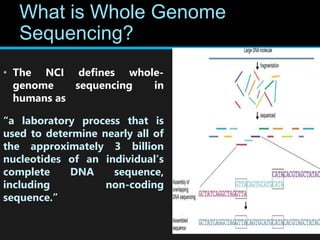

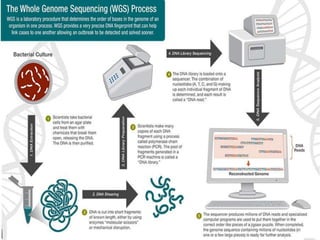

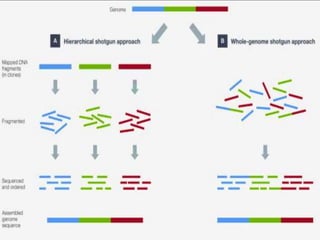
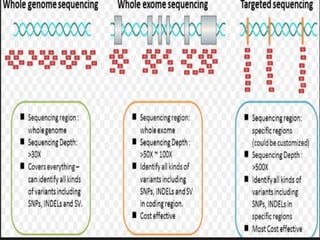







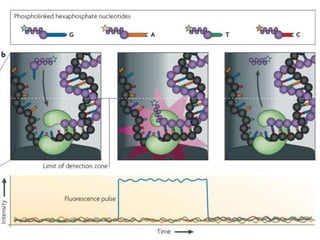
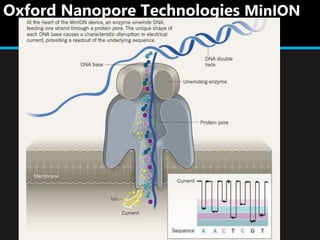

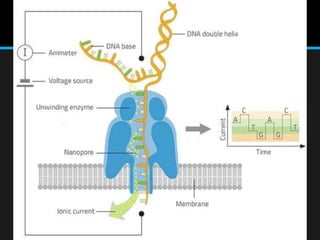



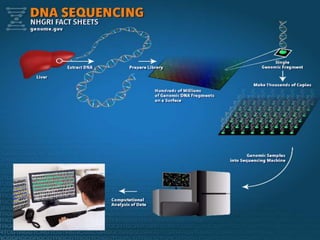



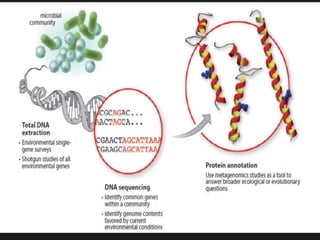
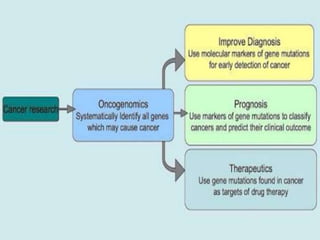
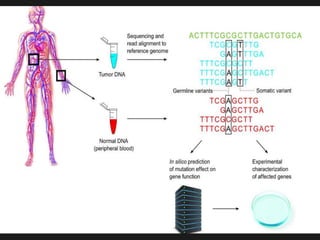

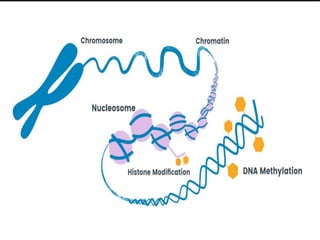



Gene sequencing is the technique that determines the order of nucleotide bases in DNA. It allows researchers to read genetic information and understand genes. The first genome sequenced was a bacteriophage in 1977. Techniques have advanced from Sanger sequencing to second-generation sequencing using platforms like Illumina and third-generation single-molecule techniques. Gene sequencing has various applications in medicine, forensics, agriculture, cancer research and more. It is an important tool for understanding genomes and their relationship to traits and disease.











































































