





















![ANALYTICAL METHODS
• In the case of AMS, [14C] is the most useful isotope for drug
metabolism studies whereas for PET [11C] is proving to be the
most useful.
• AMS is used for determining PK data by taking body samples
over time, processing the samples in the laboratory and then
analysing their drug content.
• PET provides primarily PD data through real-time imaging and
some limited PK data.](https://image.slidesharecdn.com/md-140904033534-phpapp02/85/Microdosing-Phase-0-studies-26-320.jpg)
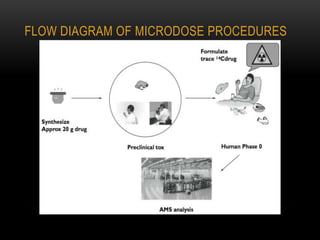


















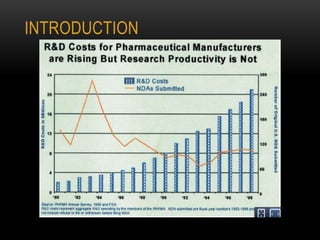
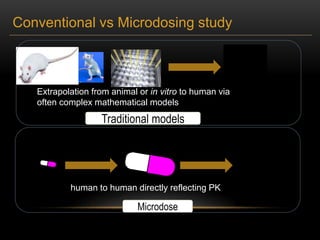
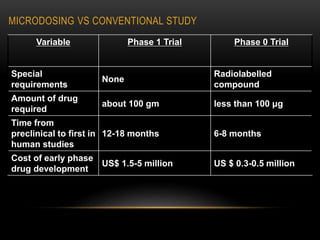
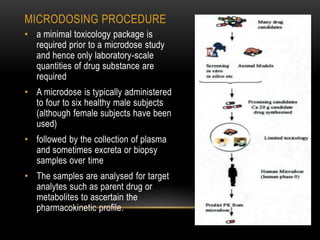
This document discusses microdosing studies, which involve administering very small, sub-therapeutic doses of drug candidates to humans early in clinical trials. The goals are to obtain human pharmacokinetic and metabolic data prior to traditional Phase 1 trials in order to select promising candidates and eliminate unsuccessful ones earlier. Microdosing studies have advantages like accelerating development timelines and reducing costs by focusing resources on candidates more likely to succeed in later trials. The document covers the concept, goals, procedures, uses, advantages, and regulatory guidelines of microdosing studies.


















![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)
















































































