





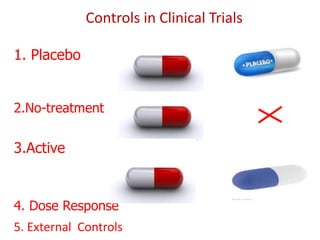
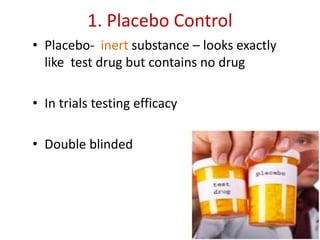












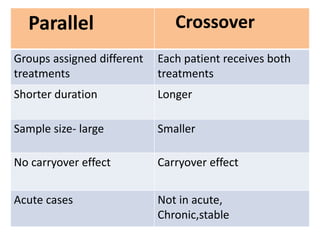
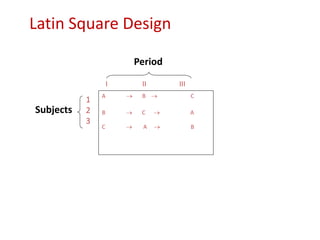



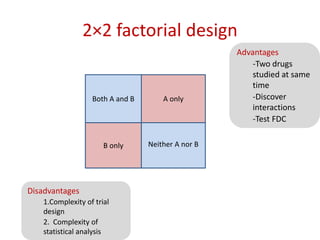
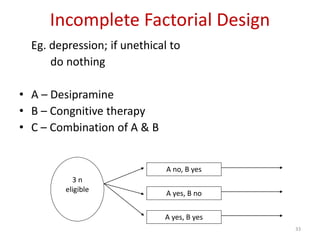

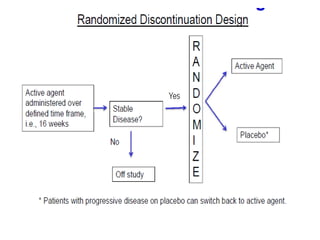
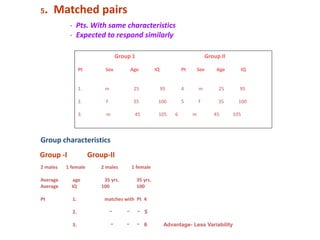







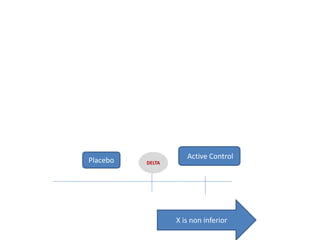


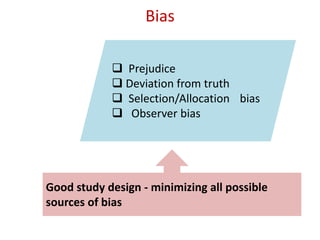









![Blinding not possible when
• Surgery with non–surgical treatment
• Types of dialysis [hemodialysis versus
peritoneal dialysis]](https://image.slidesharecdn.com/clinicaltrialdesign-131204153142-phpapp02/85/Clinical-trial-design-57-320.jpg)



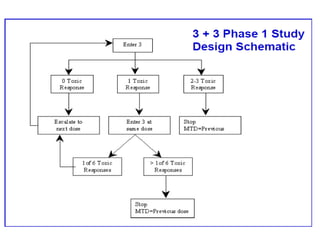
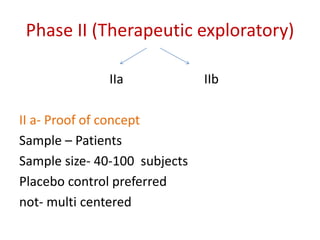
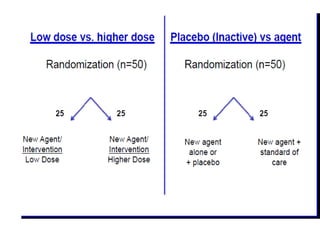


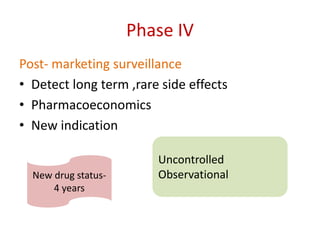

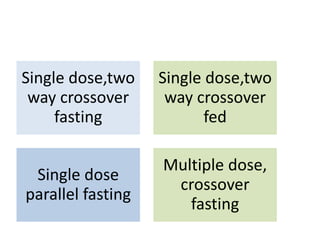

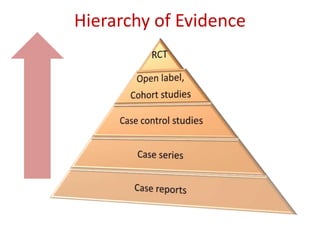





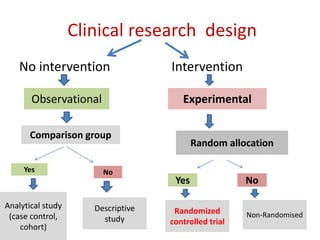
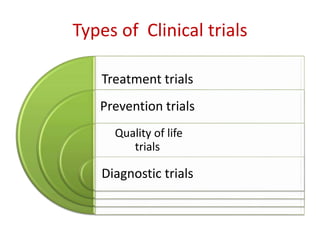
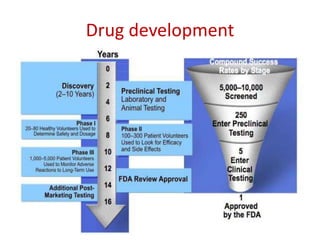
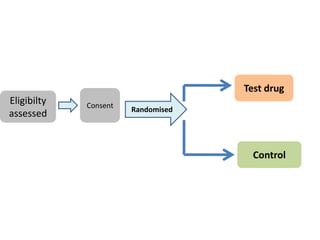
This document discusses various clinical trial designs including parallel, crossover, factorial, and adaptive designs. It describes key elements of clinical trial methodology such as randomization, blinding, placebos, and controls. The document also outlines how clinical trial designs are applied differently in each phase of drug development from Phase 0 microdosing to Phase III confirmatory trials. Key challenges in clinical trial design like controlling bias and complex statistical analysis of factorial designs are also summarized.























































































