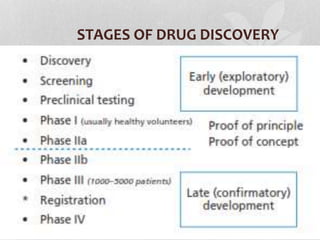
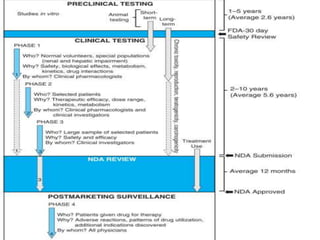
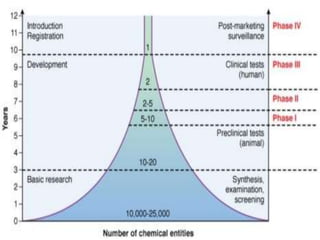
Clinical trials involve testing investigational drugs or treatments on human subjects to determine safety and efficacy. They progress through several phases, beginning with small pre-clinical trials on animals. Phase 1 trials involve 20-50 healthy volunteers to assess pharmacokinetics and safety. Phase 2 trials enroll 50-300 patient volunteers to further evaluate safety and dosage. Phase 3 trials are large randomized controlled trials of 250-1000+ subjects comparing the investigational treatment to standard treatment or placebo. If Phase 3 is successful, the results are submitted to regulatory agencies for approval to market the new drug. Post-marketing Phase 4 trials monitor long-term safety and efficacy.






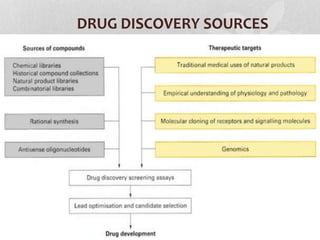



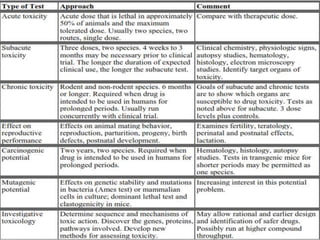

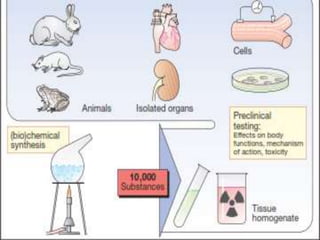










































![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)











































































