Downloaded 71 times












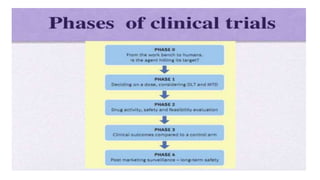

























![Phase IV Study / Clinical trial
Helps to detect rare ADRs, Drug interactions
Also to explore new uses for drugs [Sometimes called Phase V]
PERIODIC SAFETY UPDATE REPORTS
To be submitted by the manufacturer every 6 months for 2 yrs and
then annually for next 2 yrs after marketing approval](https://image.slidesharecdn.com/clincialtrialsandtypes-160926102045/85/Clincial-trials-and-types-41-320.jpg)





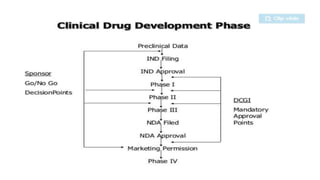
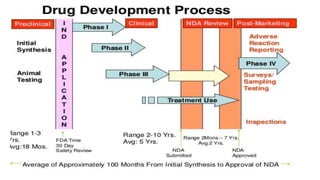
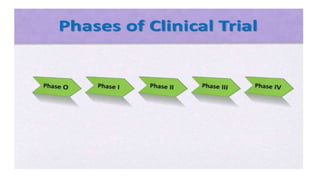
The document provides an overview of the clinical trial process for new drugs. It discusses the various phases of clinical trials including preclinical testing in animals, Phase 0 microdosing studies, Phase I safety trials in healthy volunteers, Phase II small efficacy trials in patients, and Phase III large randomized controlled trials to confirm efficacy. The goal of clinical trials is to systematically evaluate a new drug's safety and efficacy in humans at various dose levels before it can be approved for marketing. Each phase addresses different objectives and increases in size and scope as the trials progress from preclinical to final approval stages.






























































































