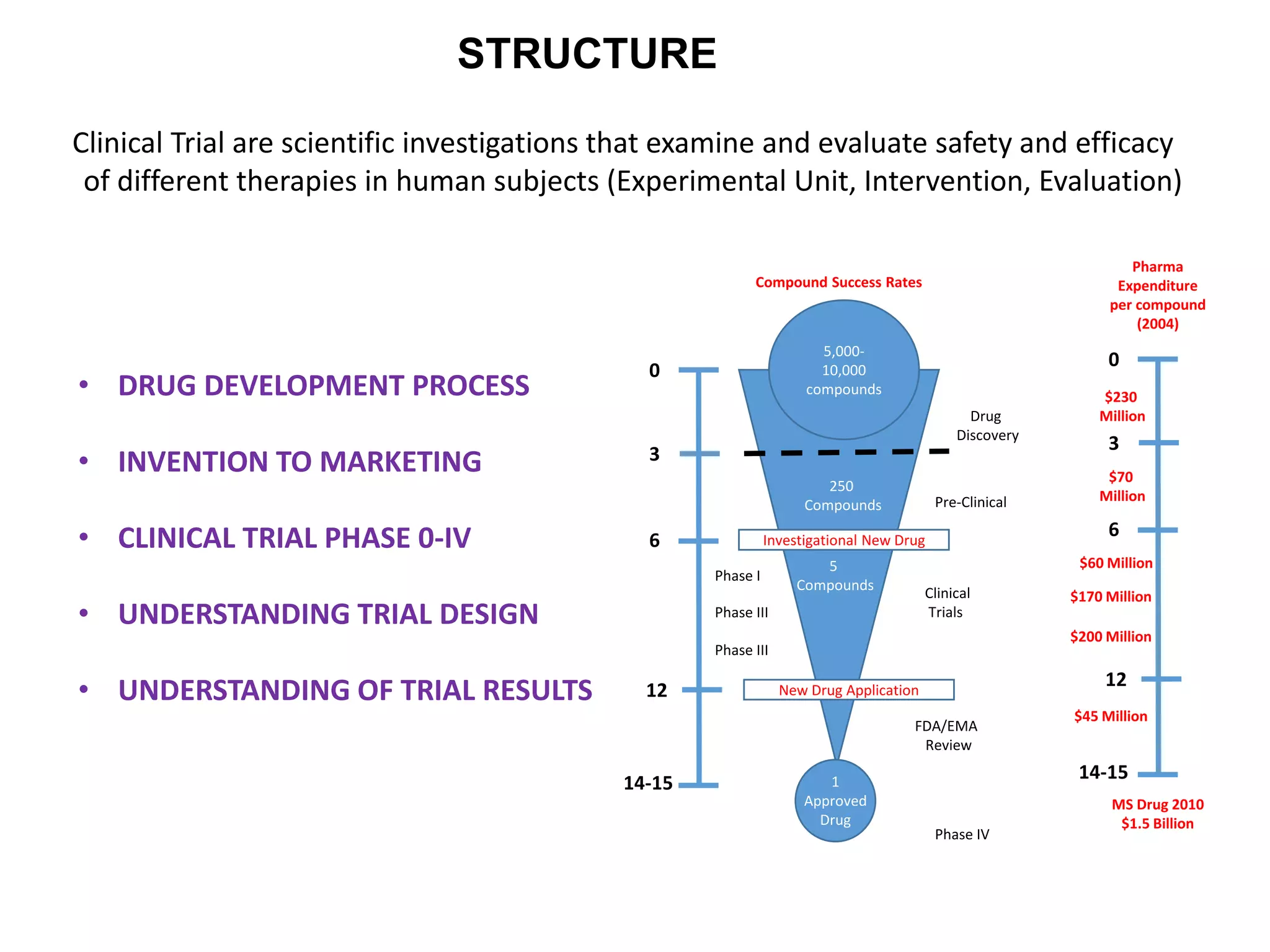
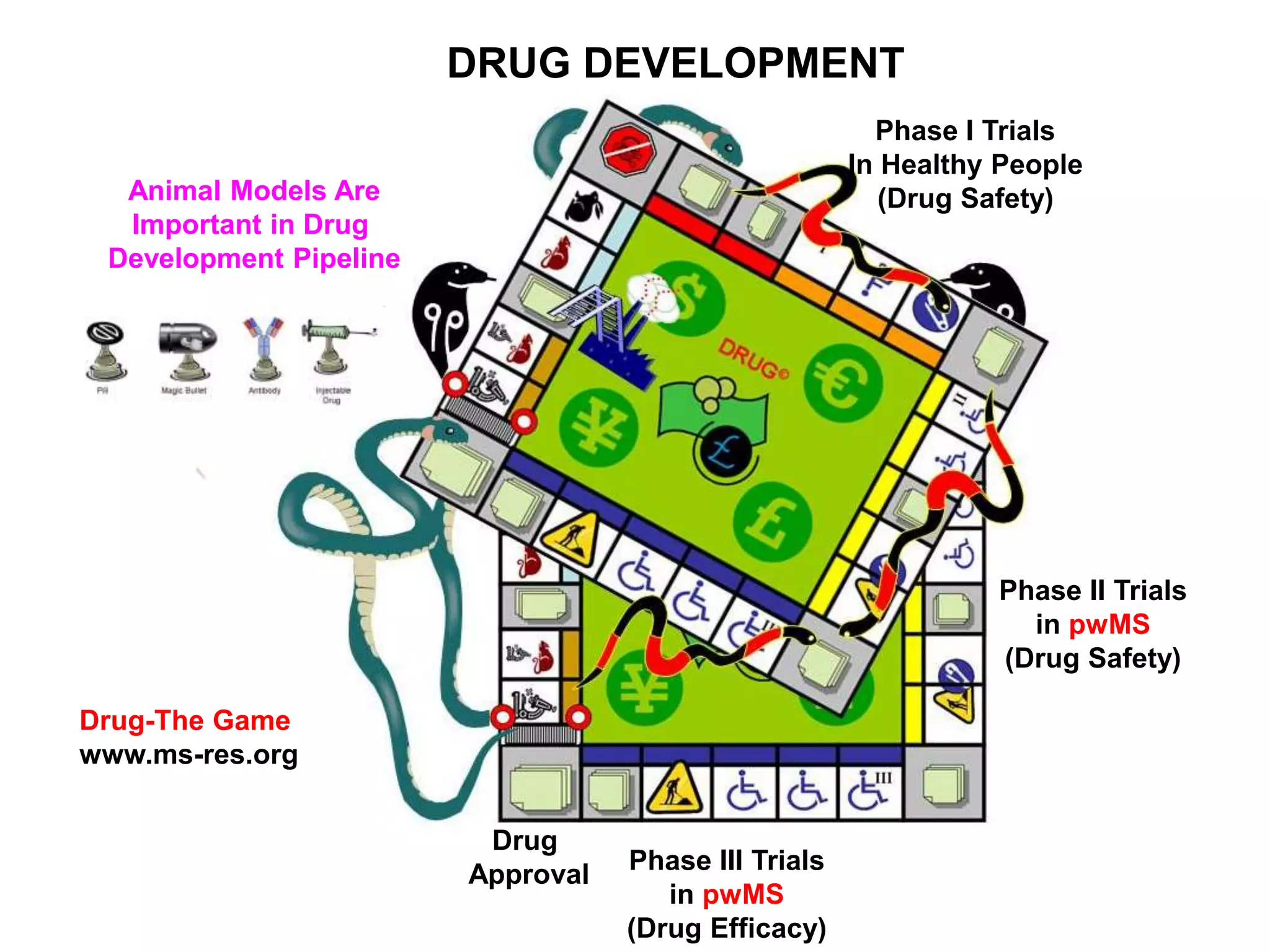
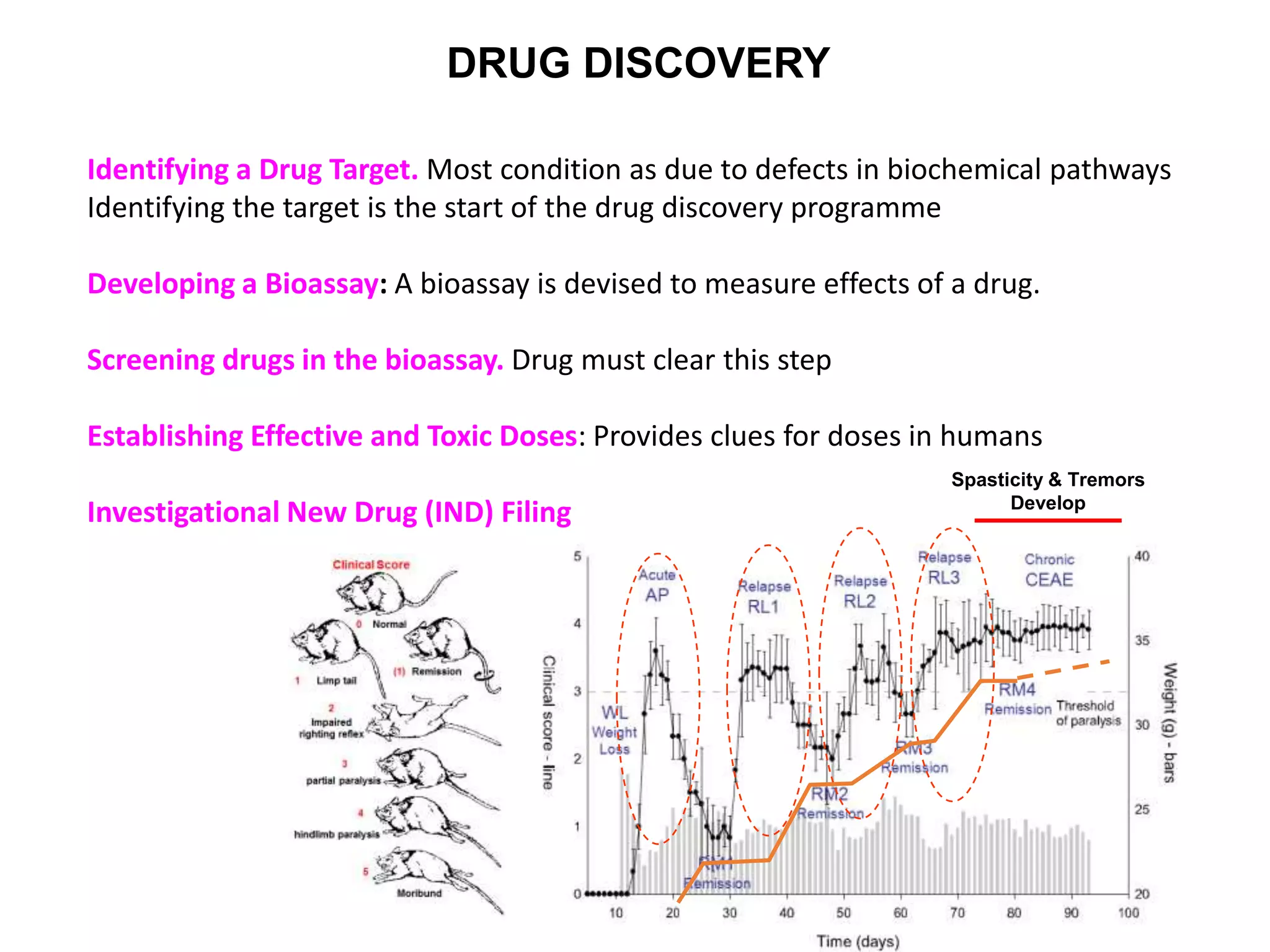
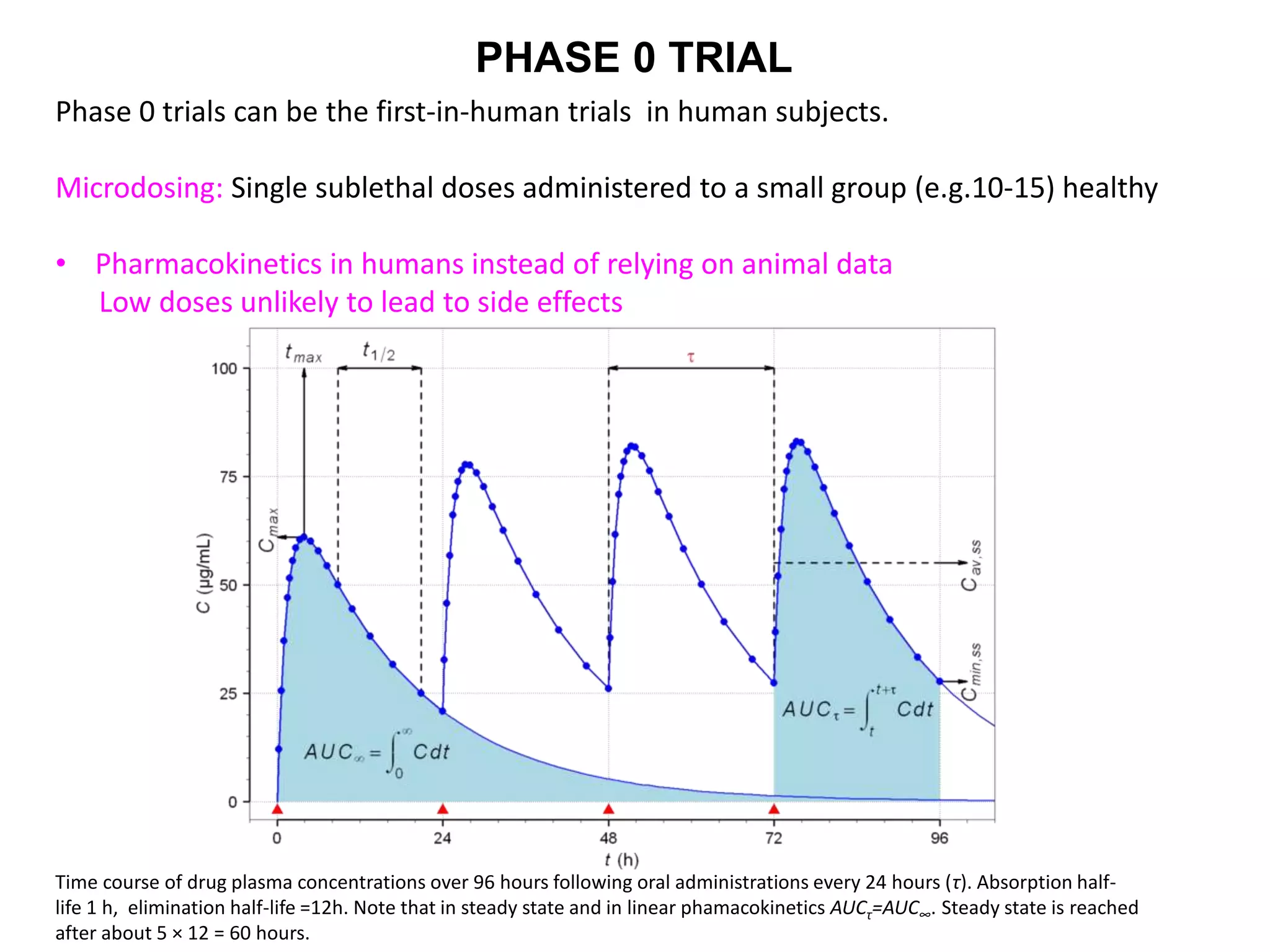
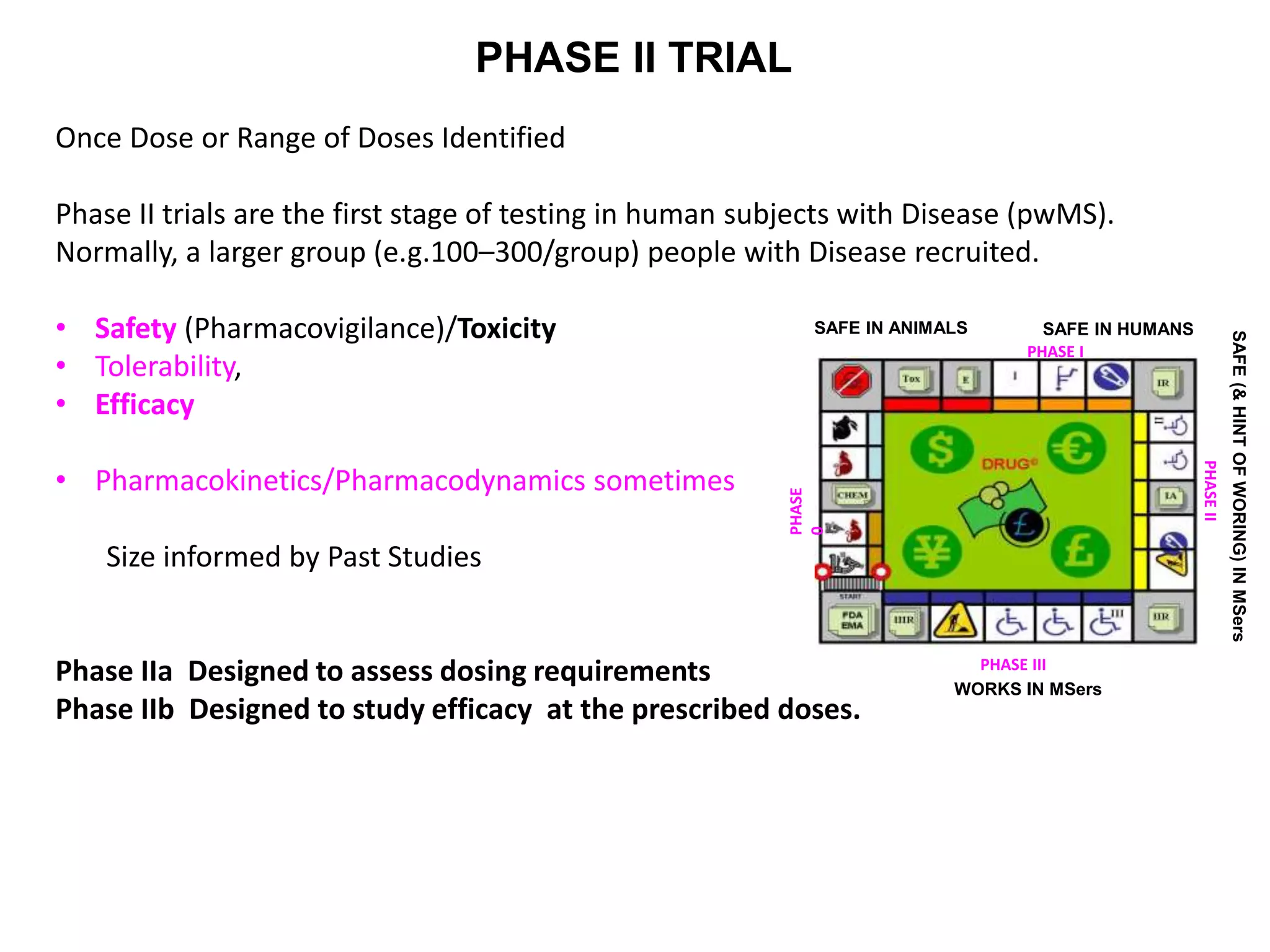
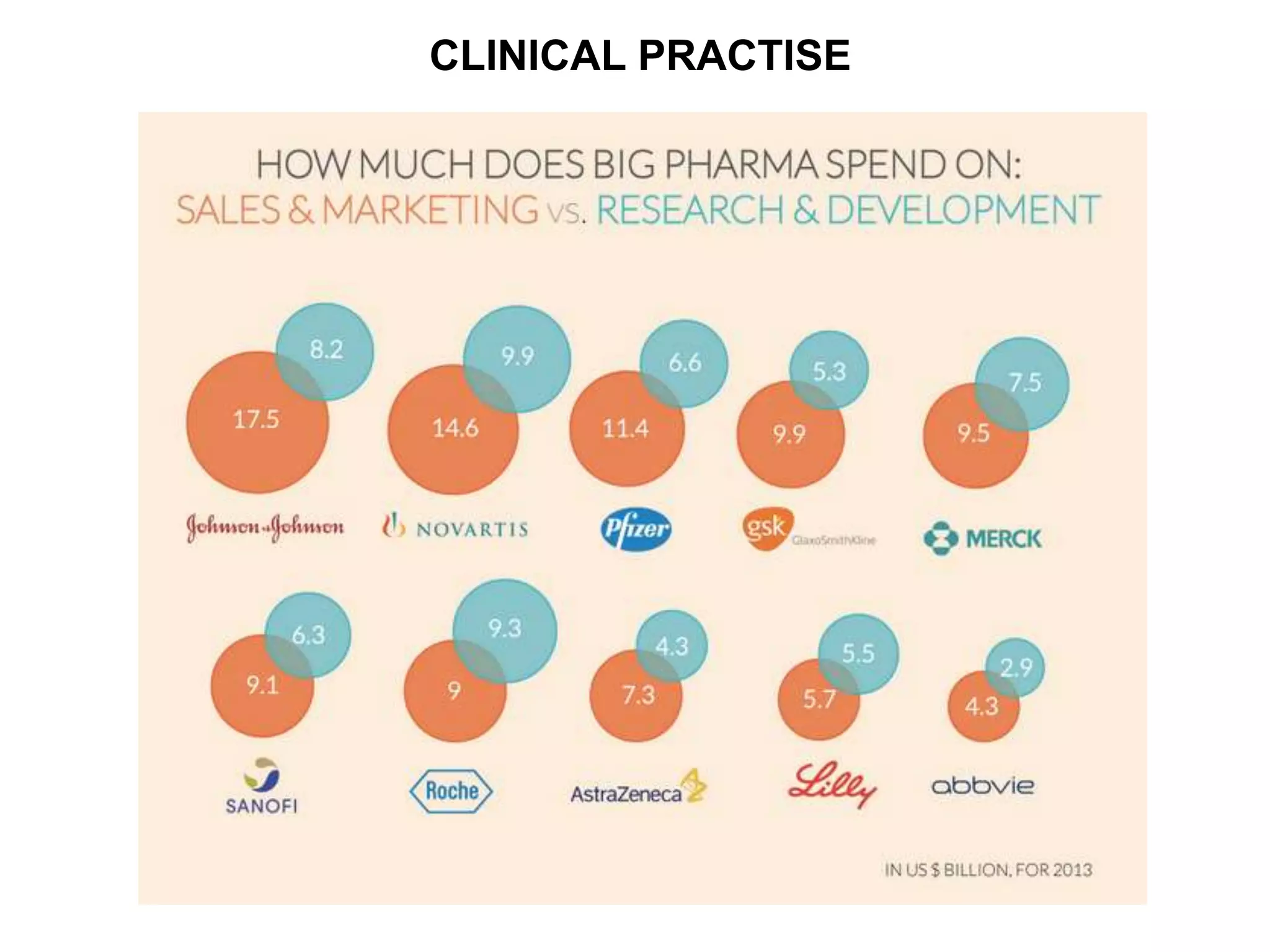
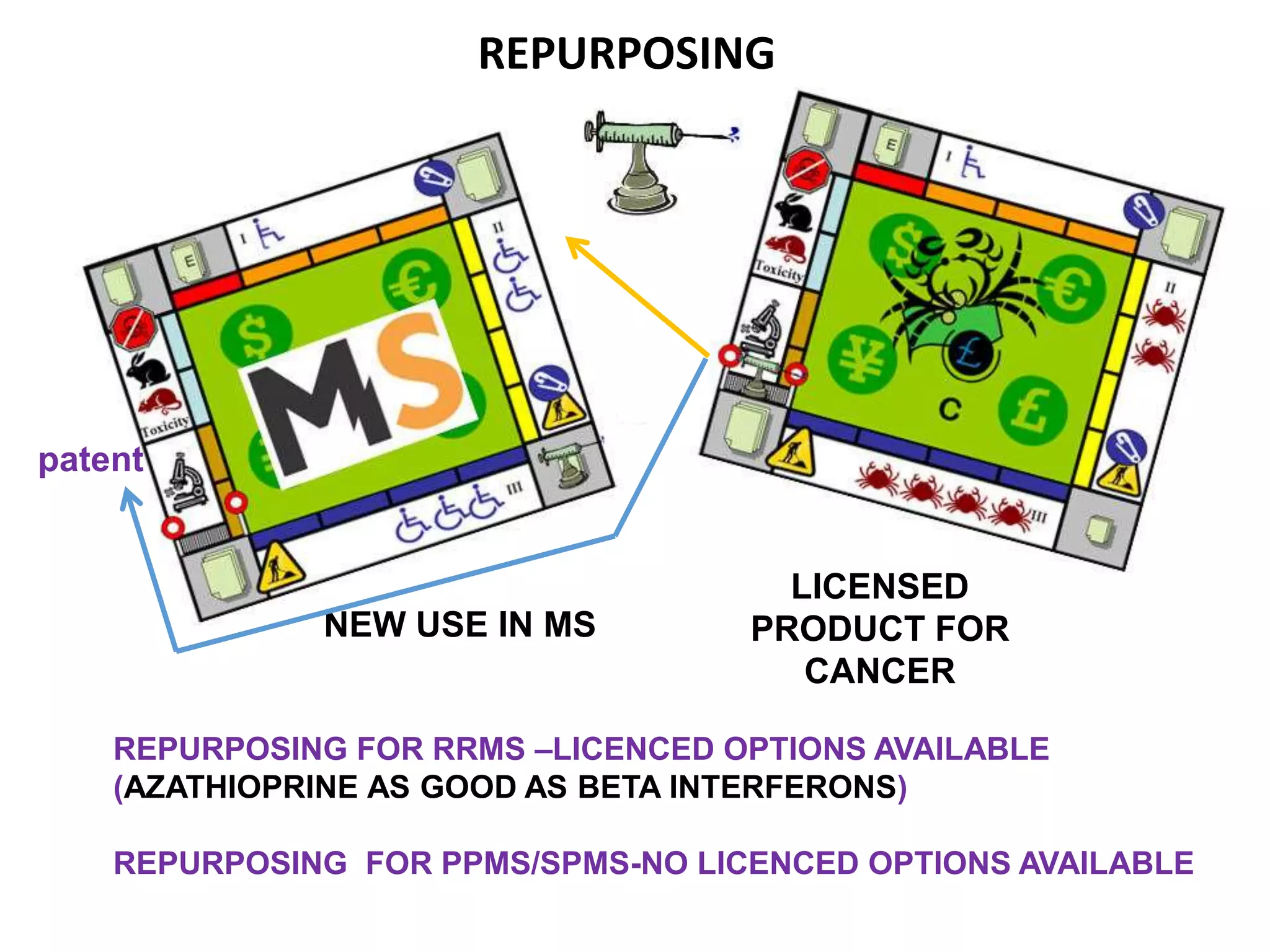
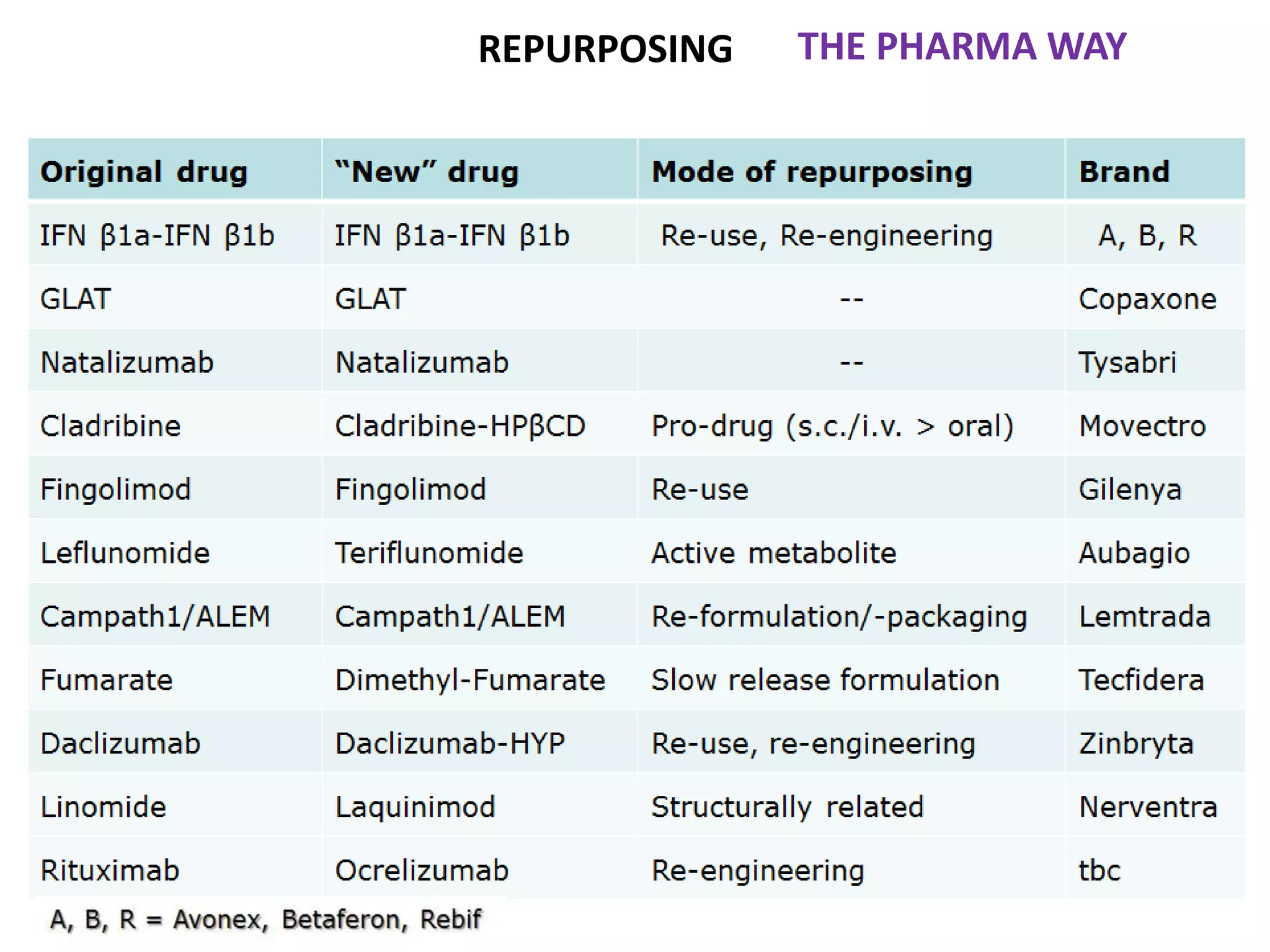
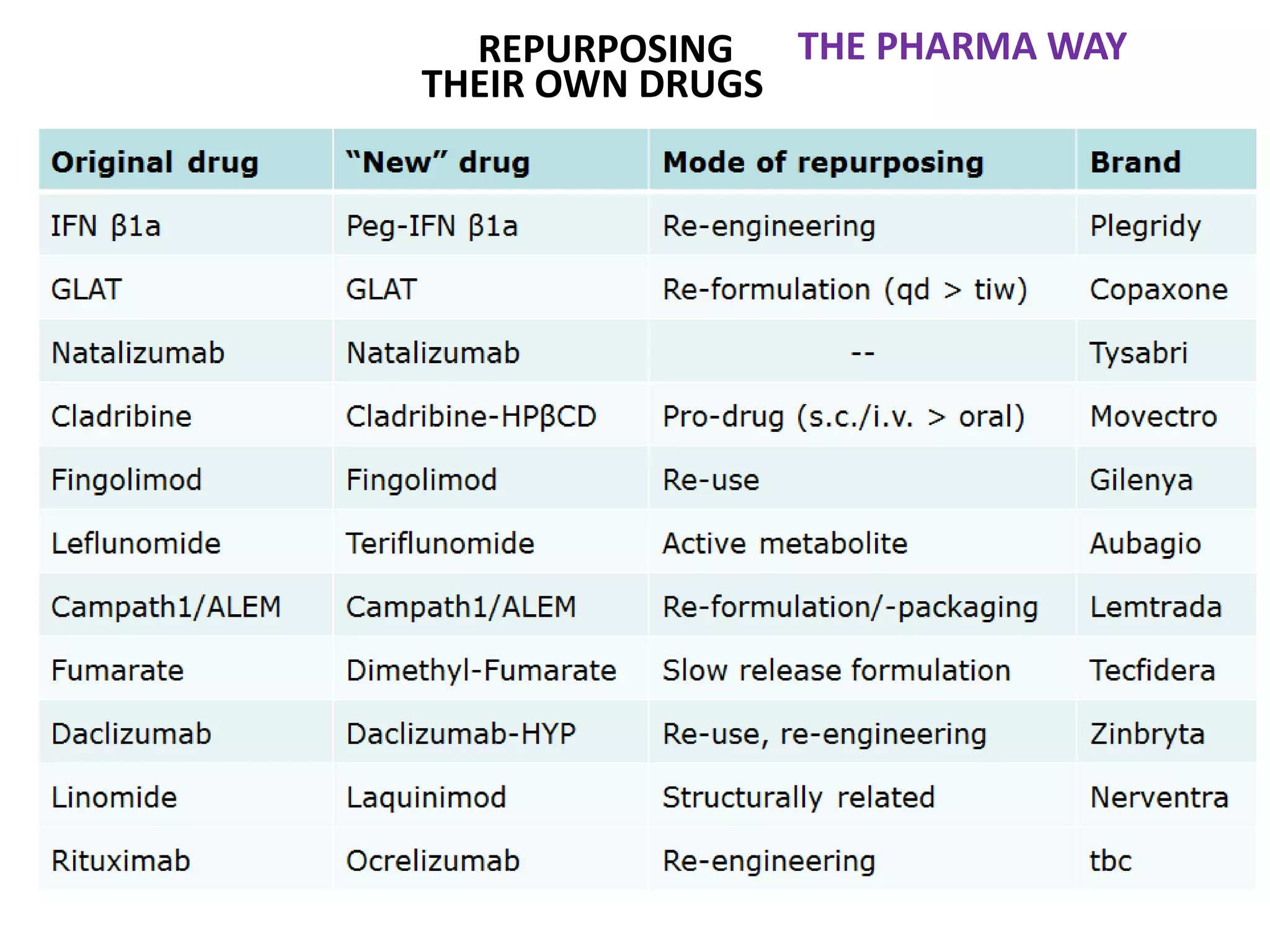
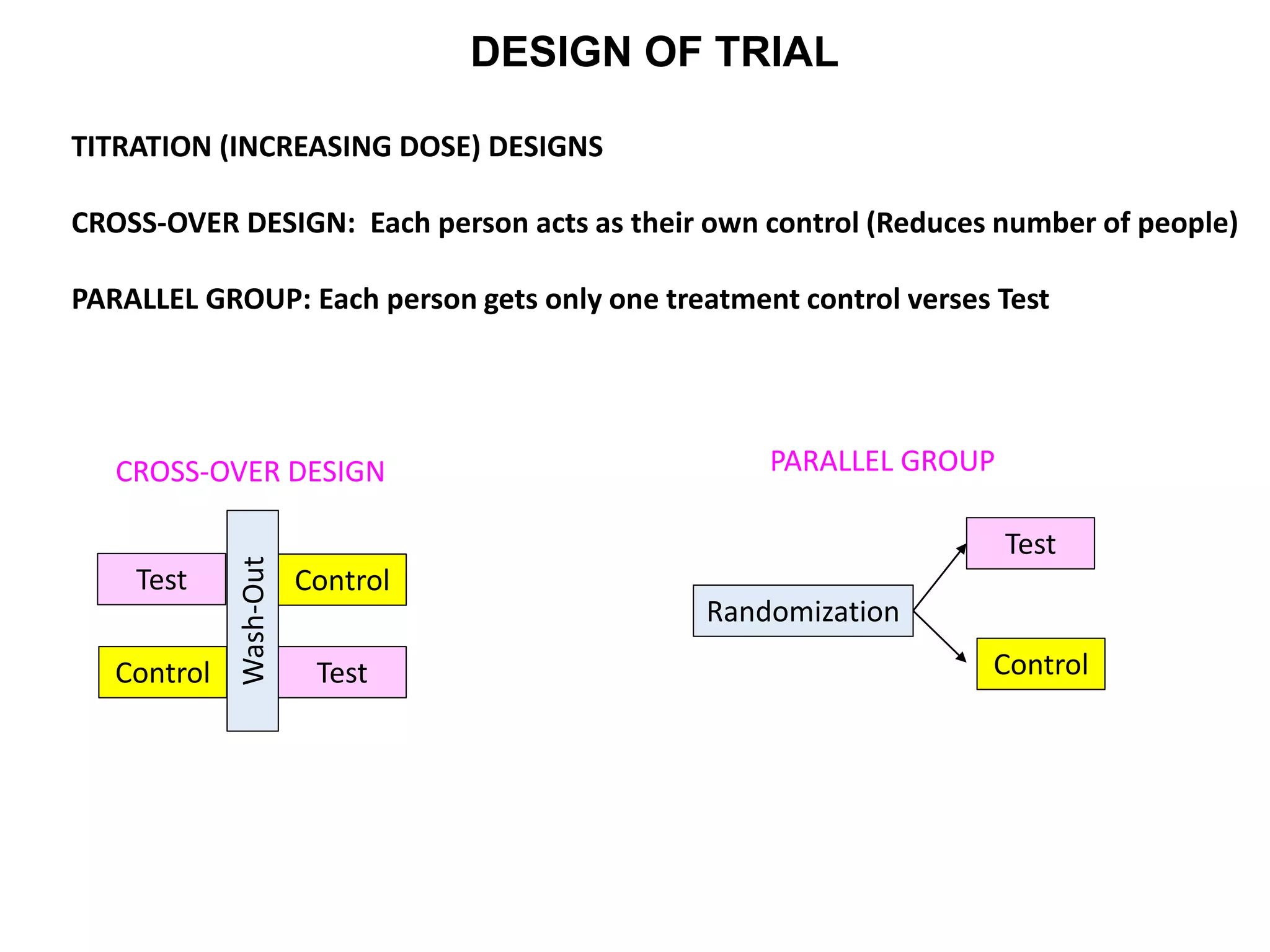
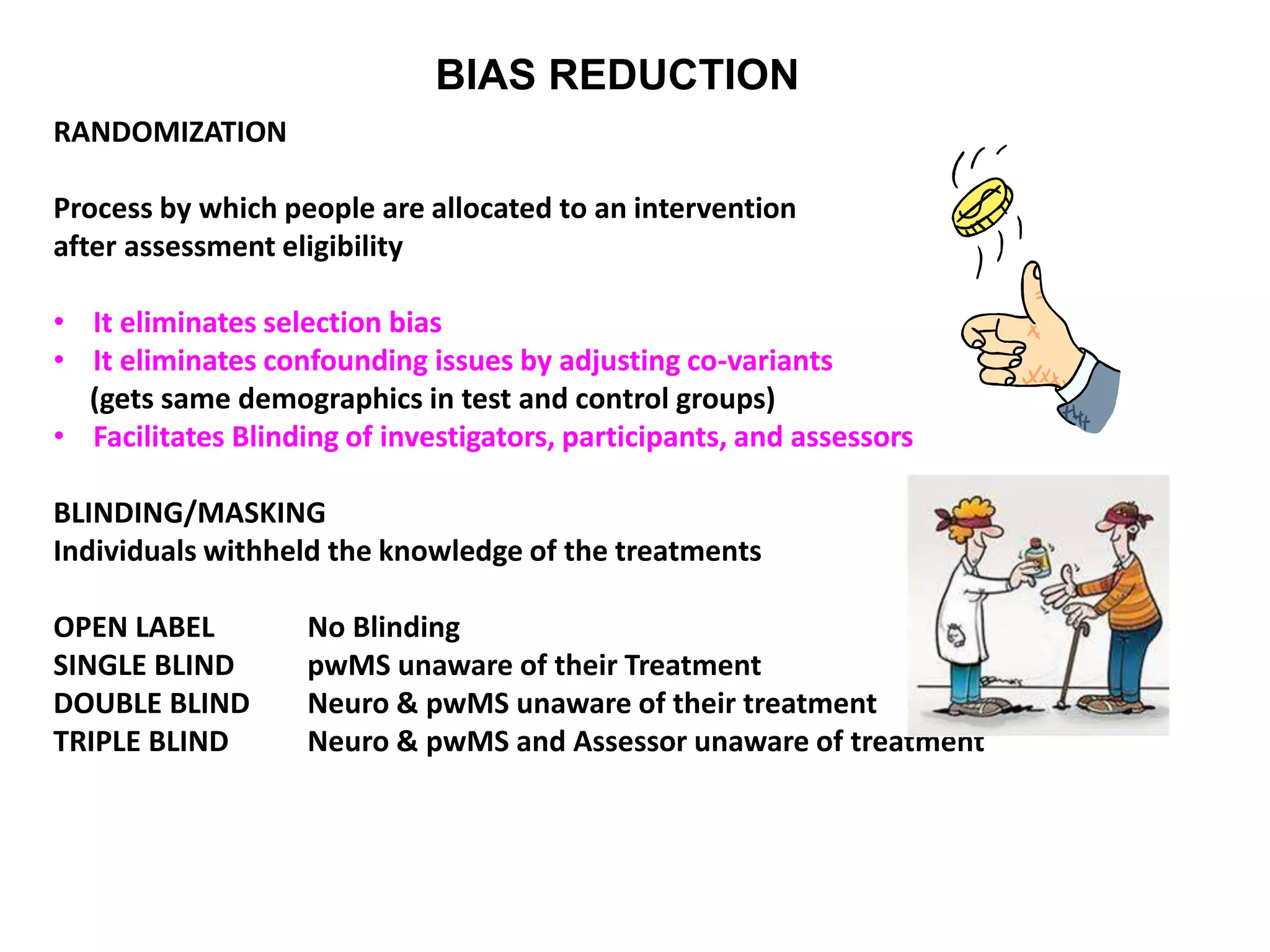
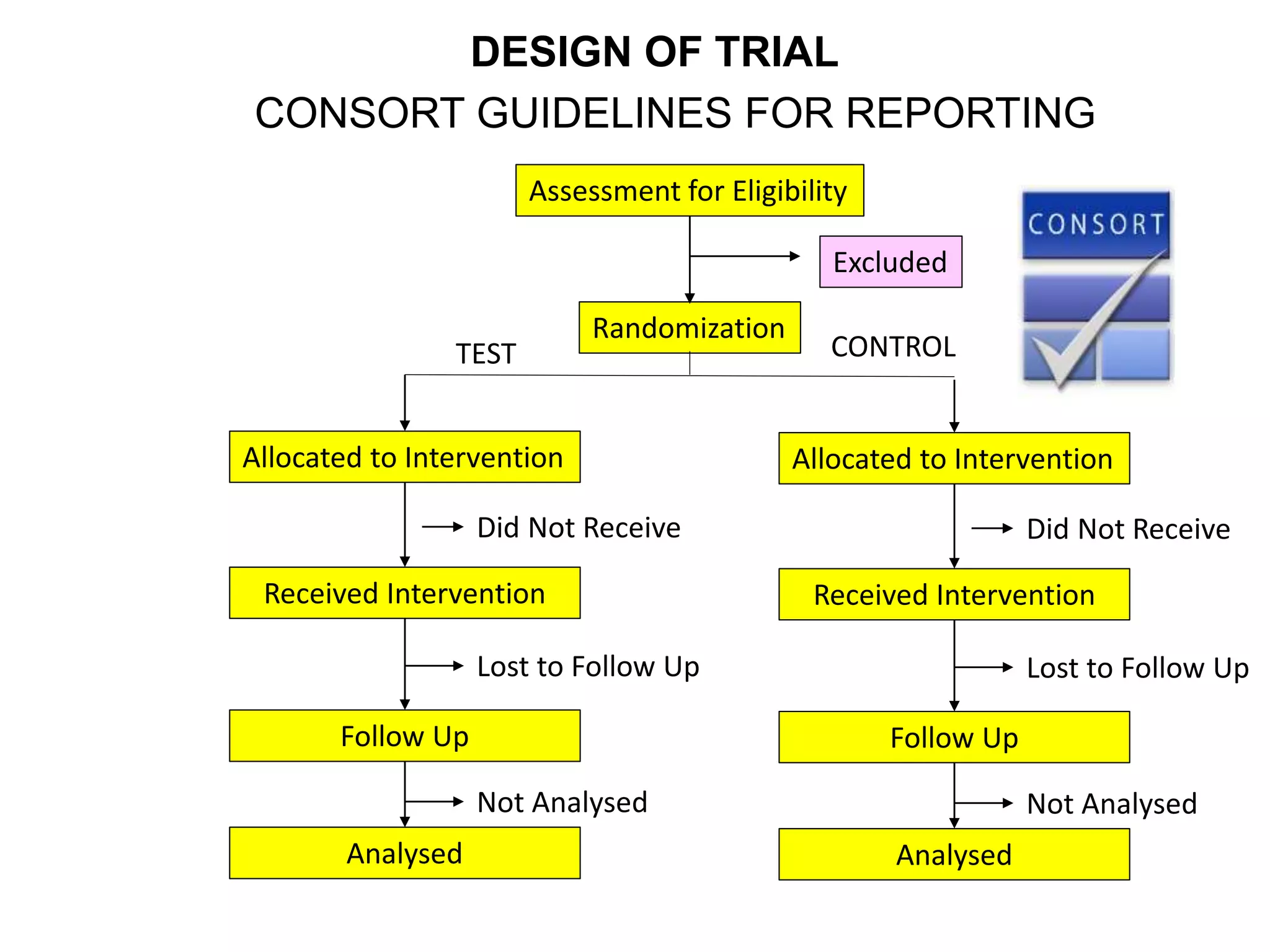
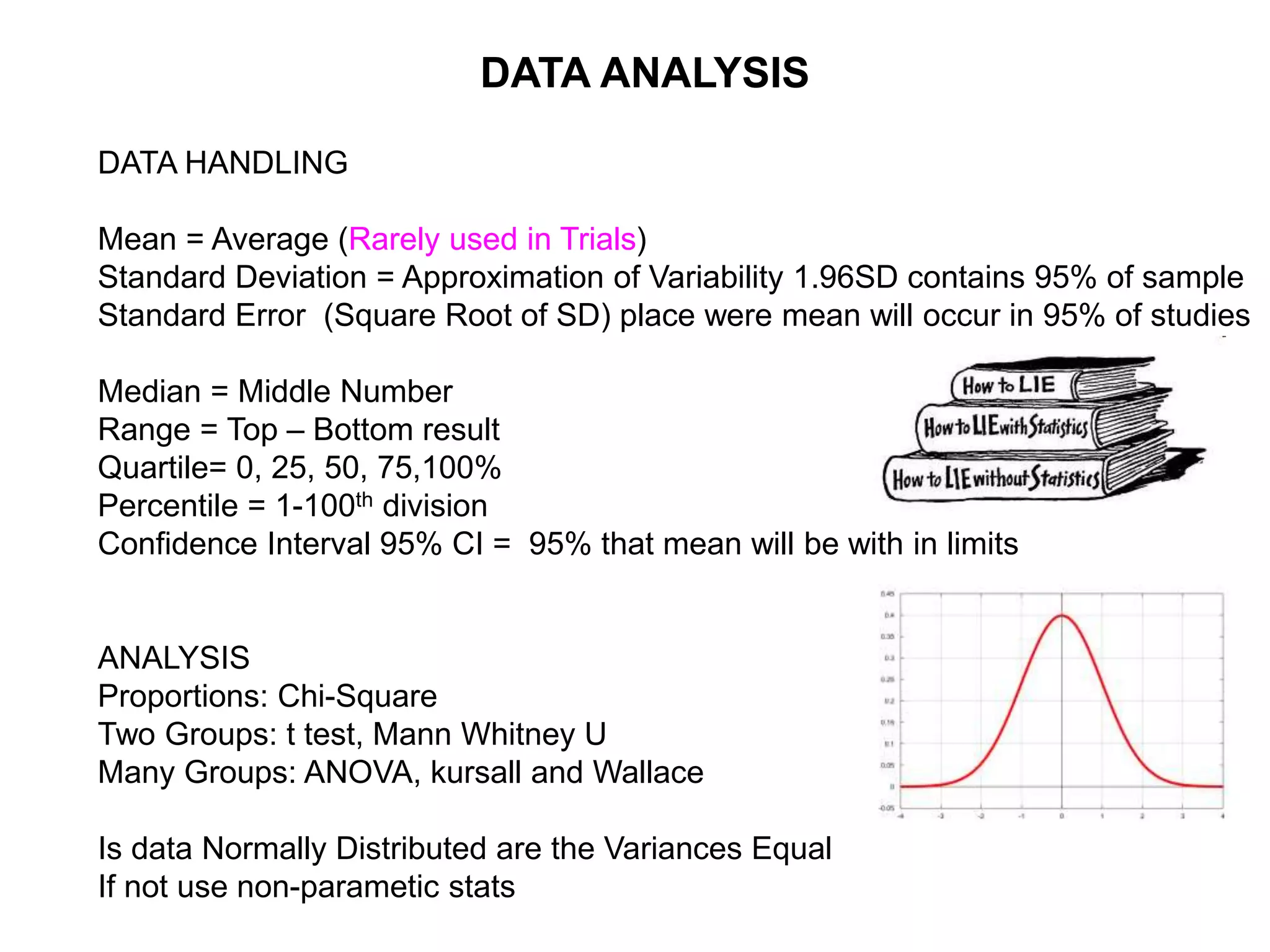
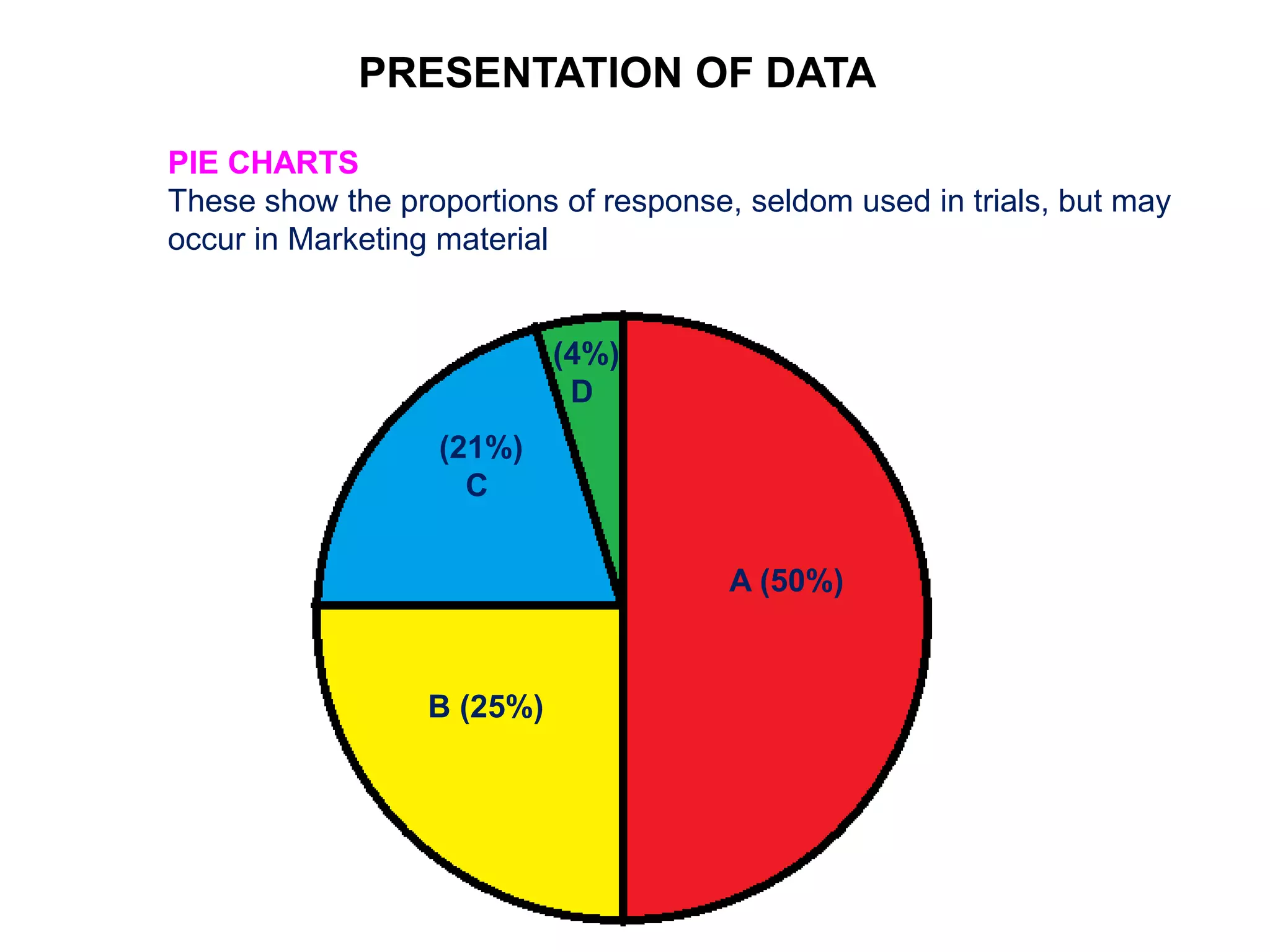
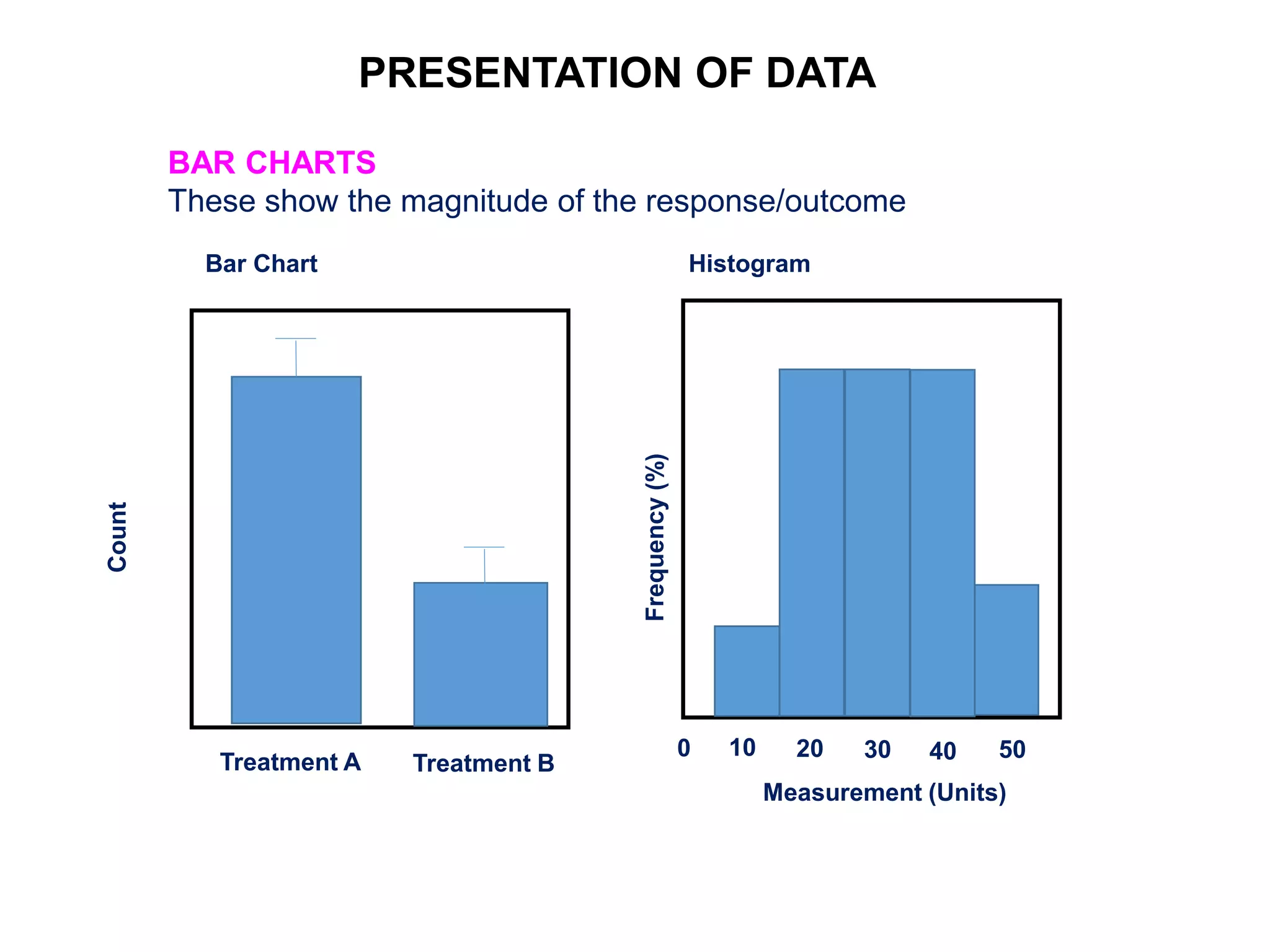
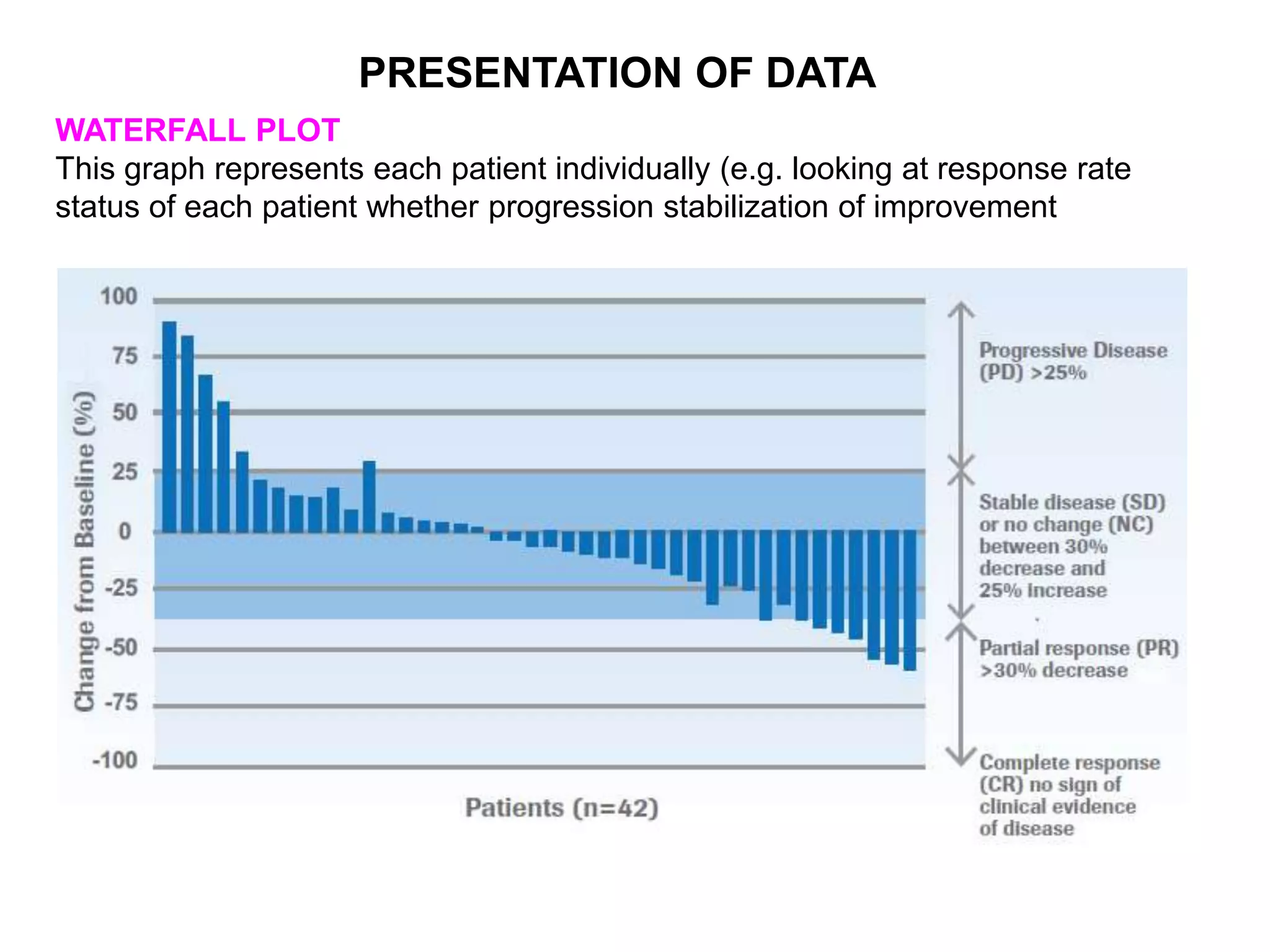
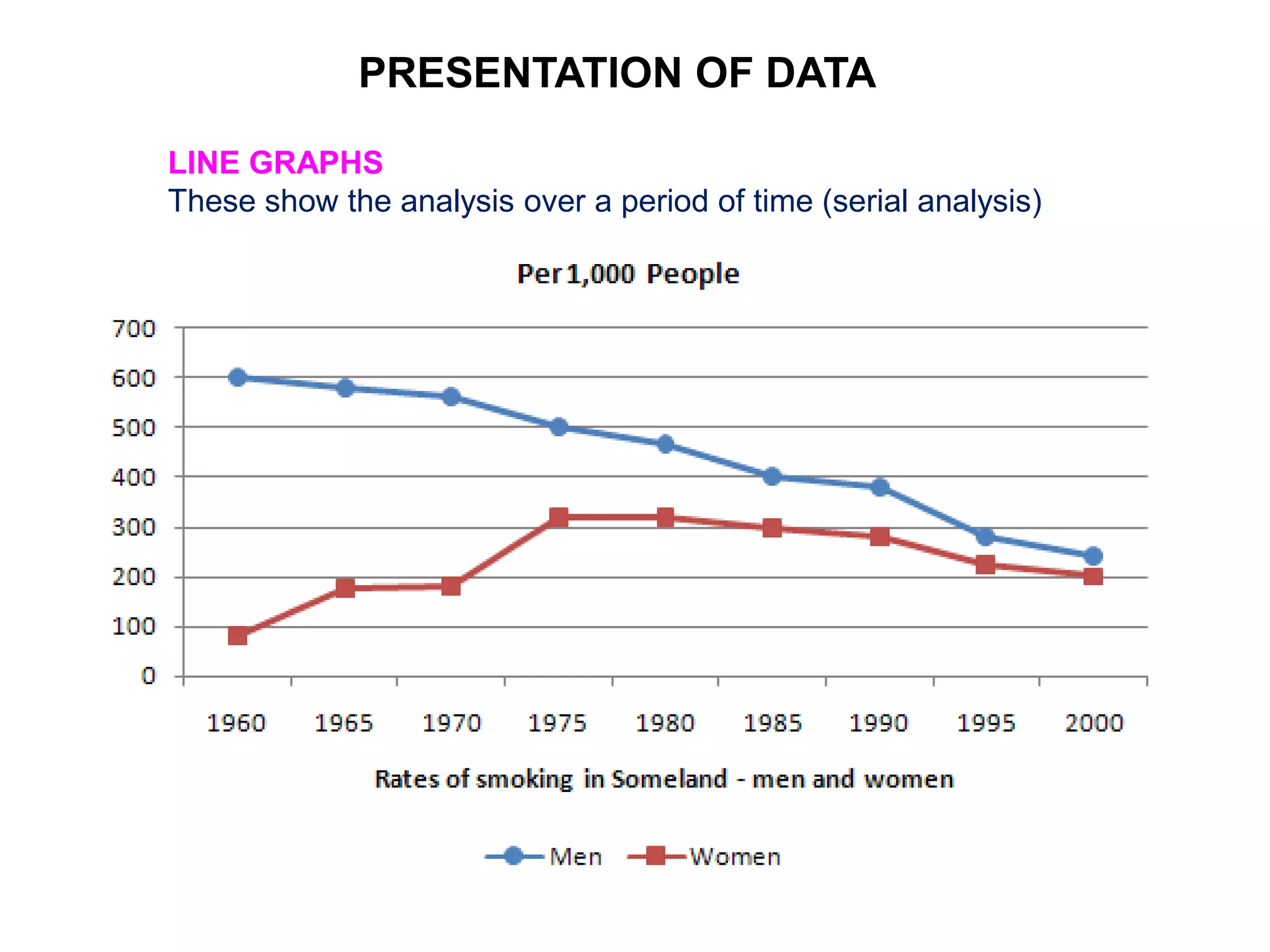
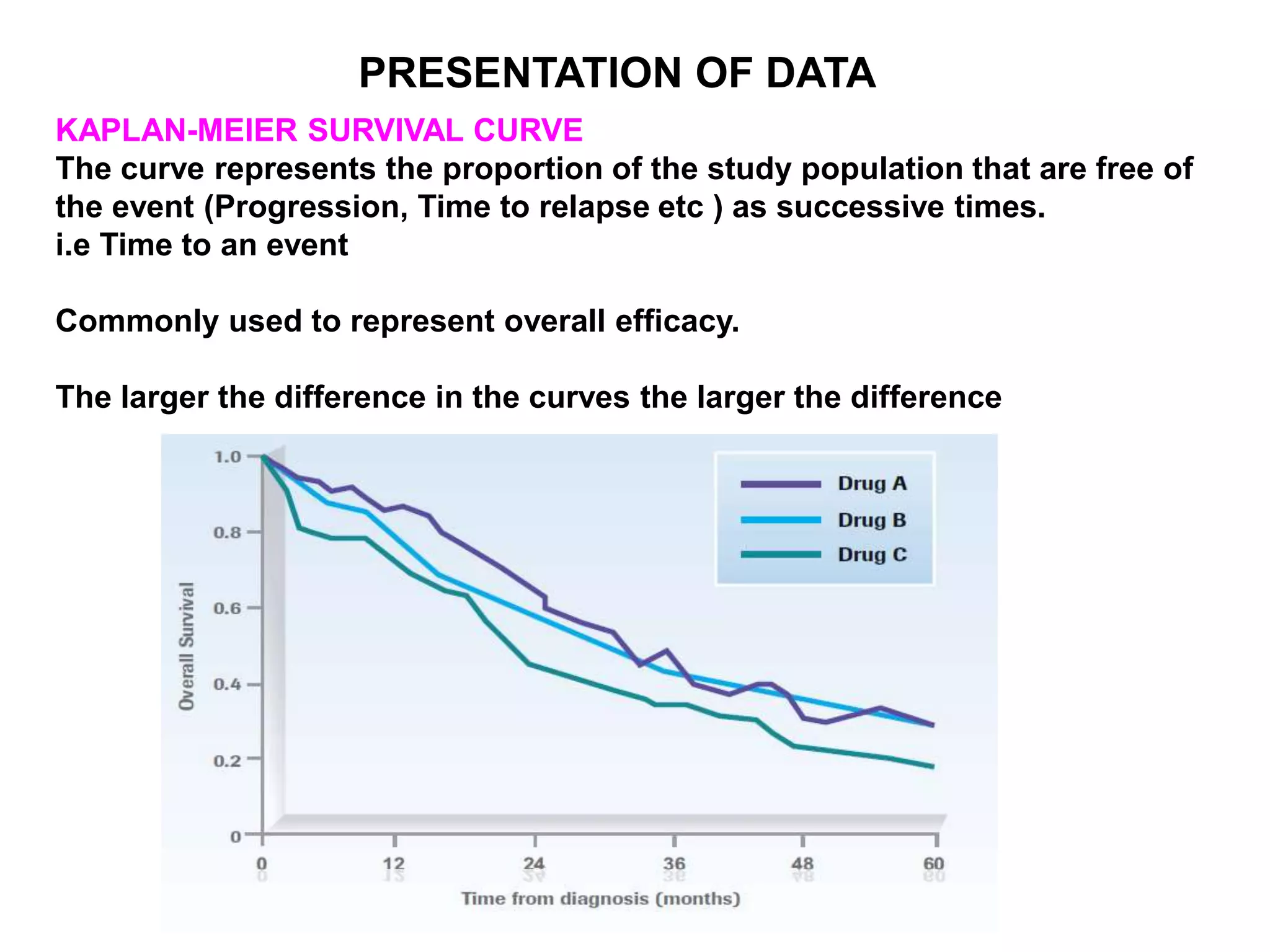
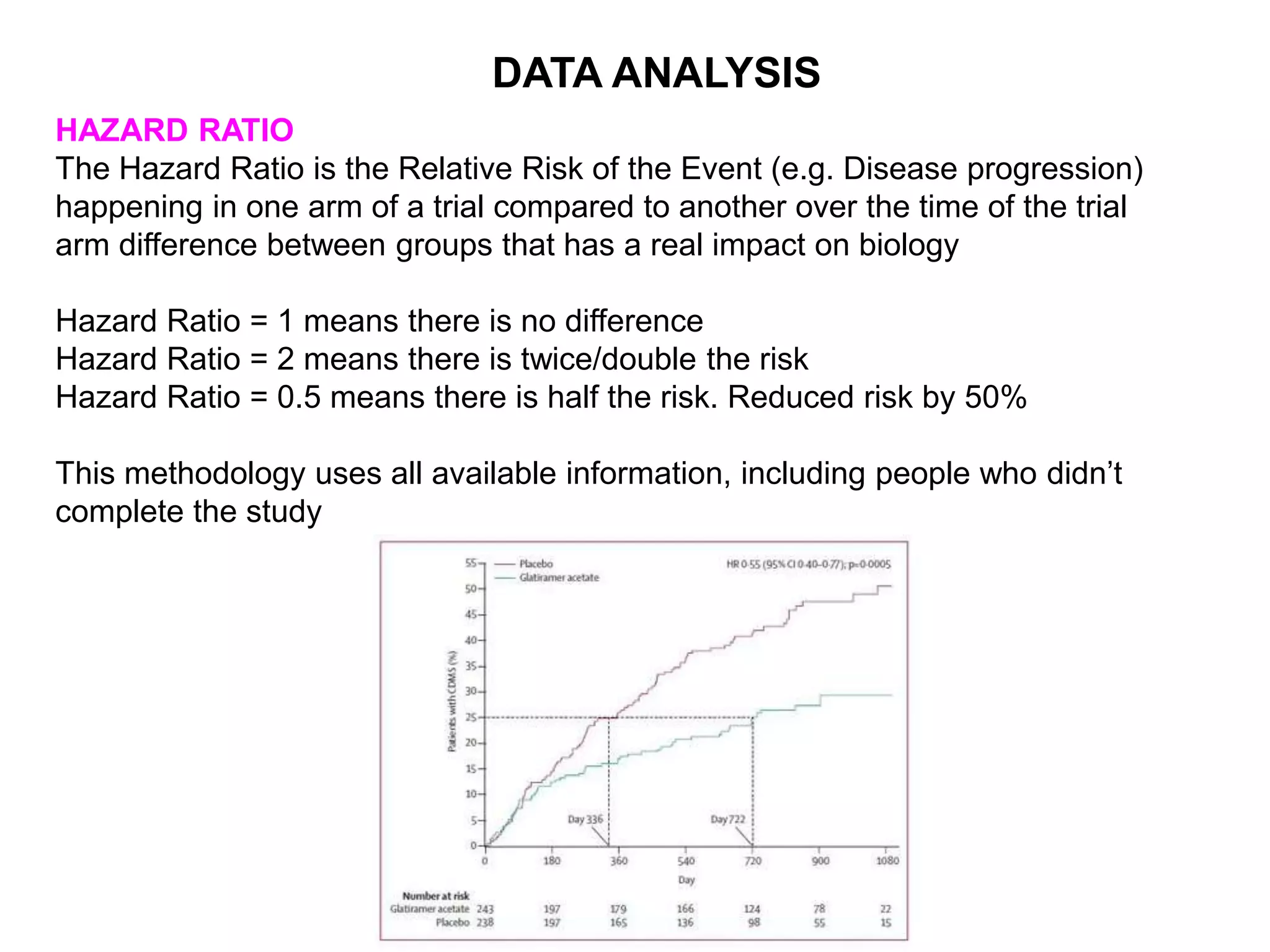
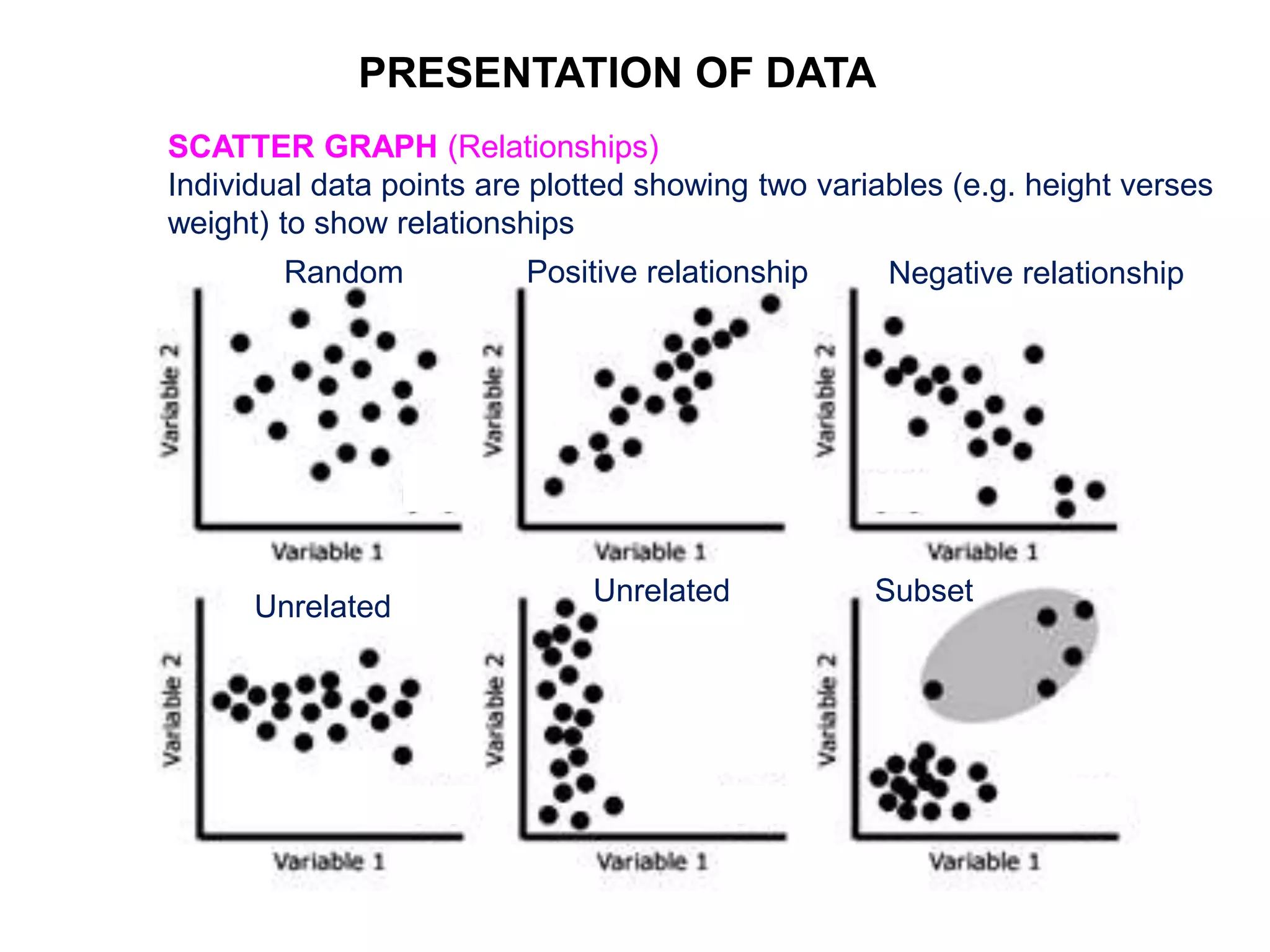
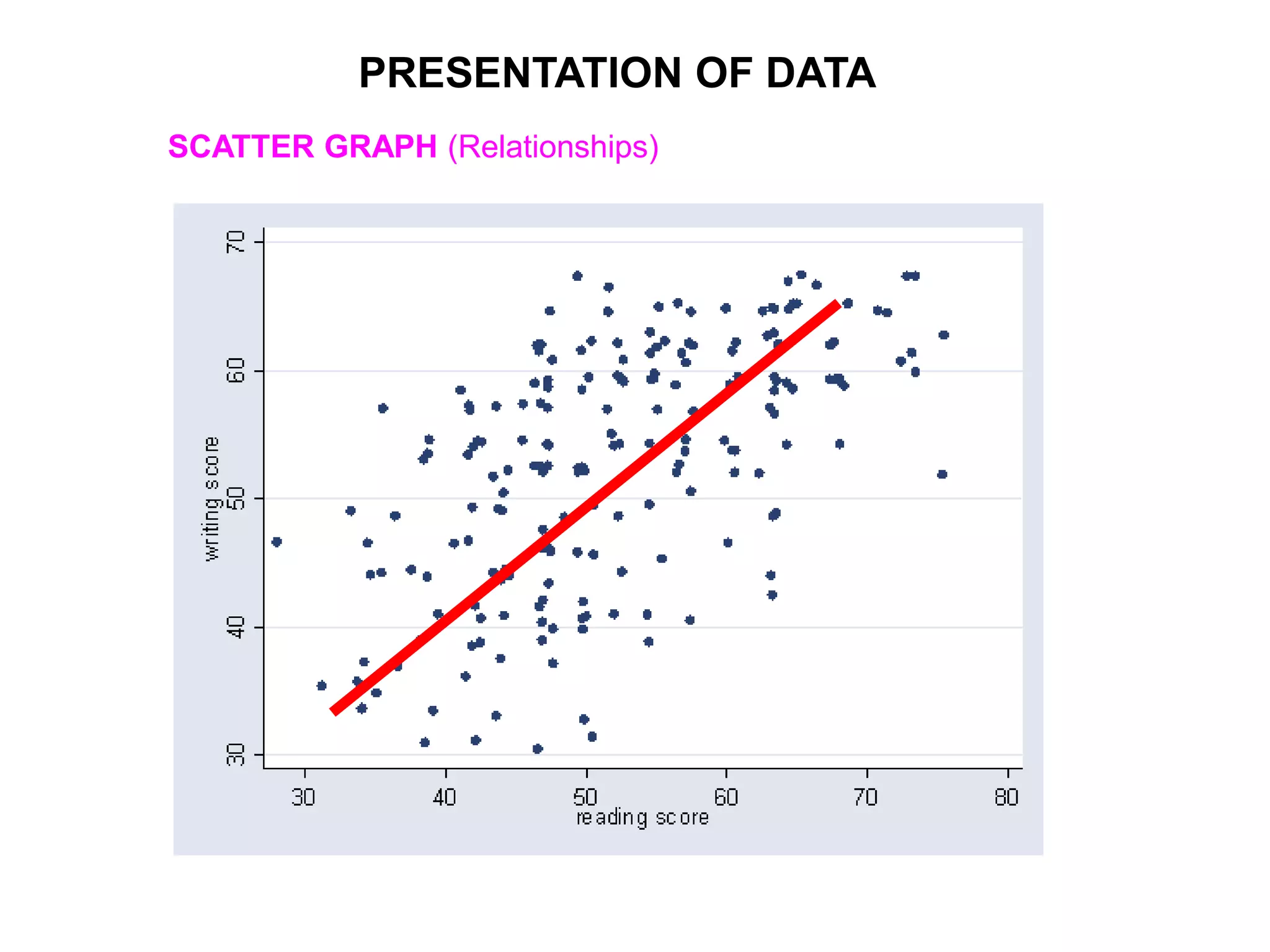
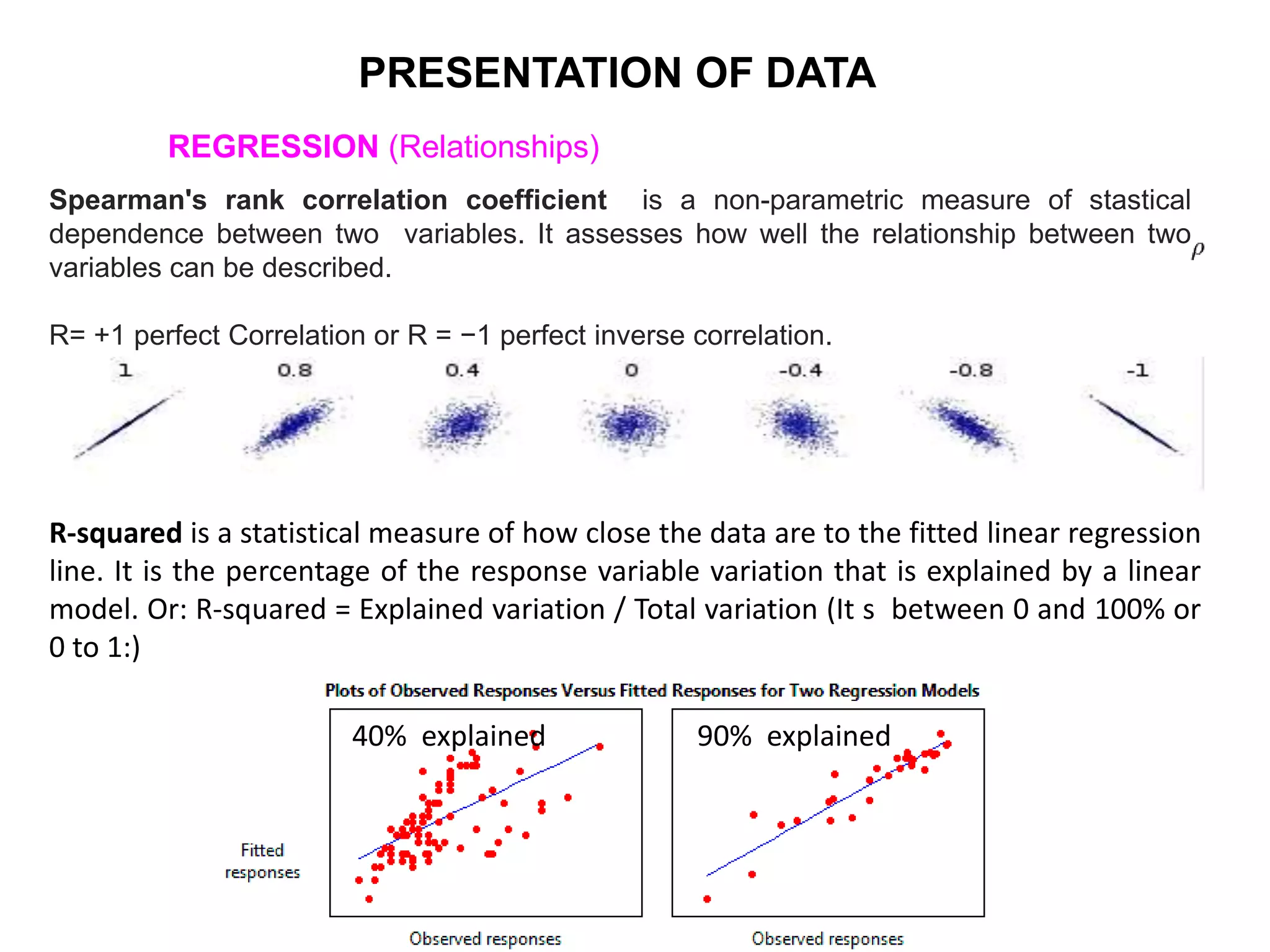
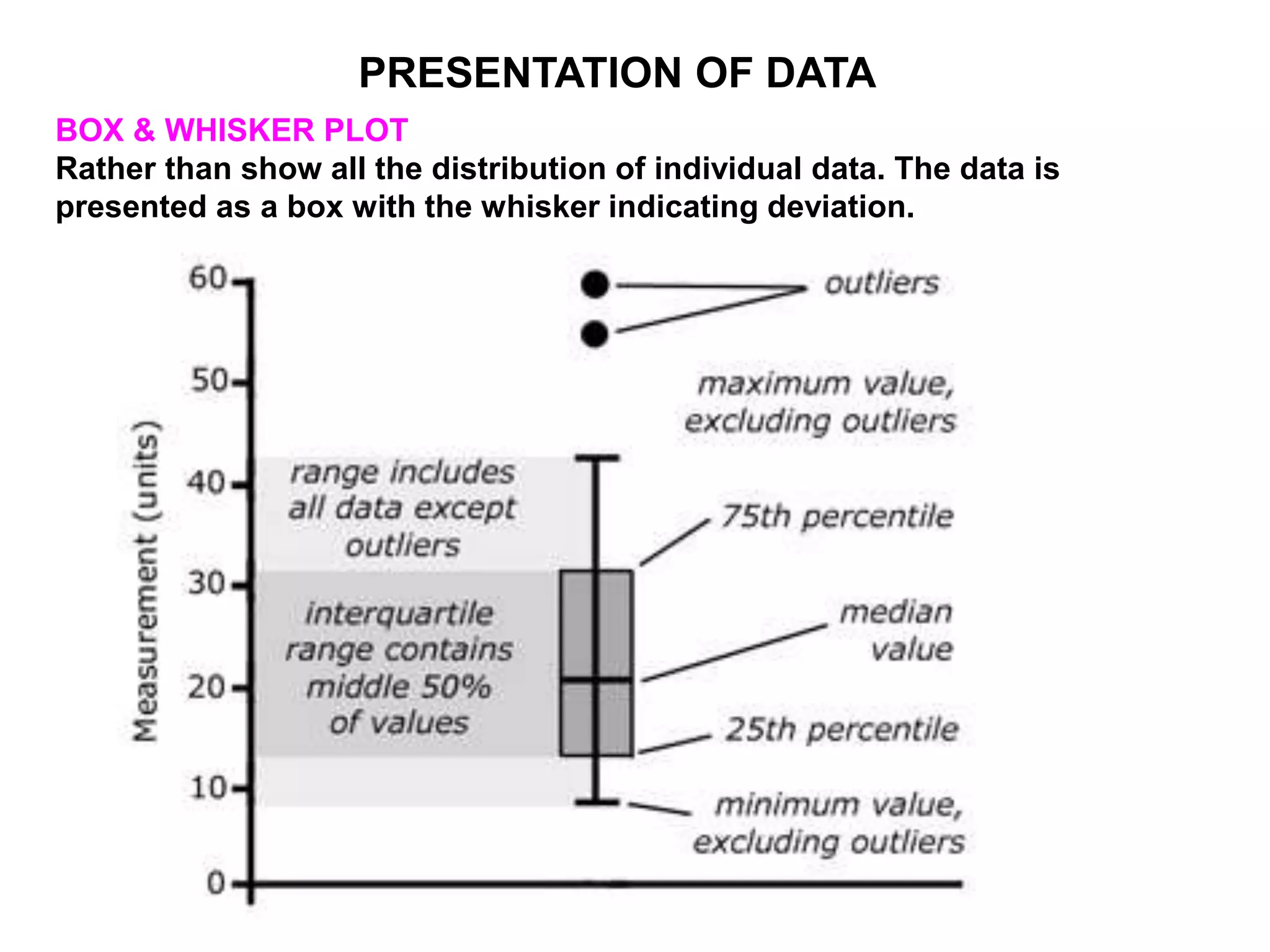
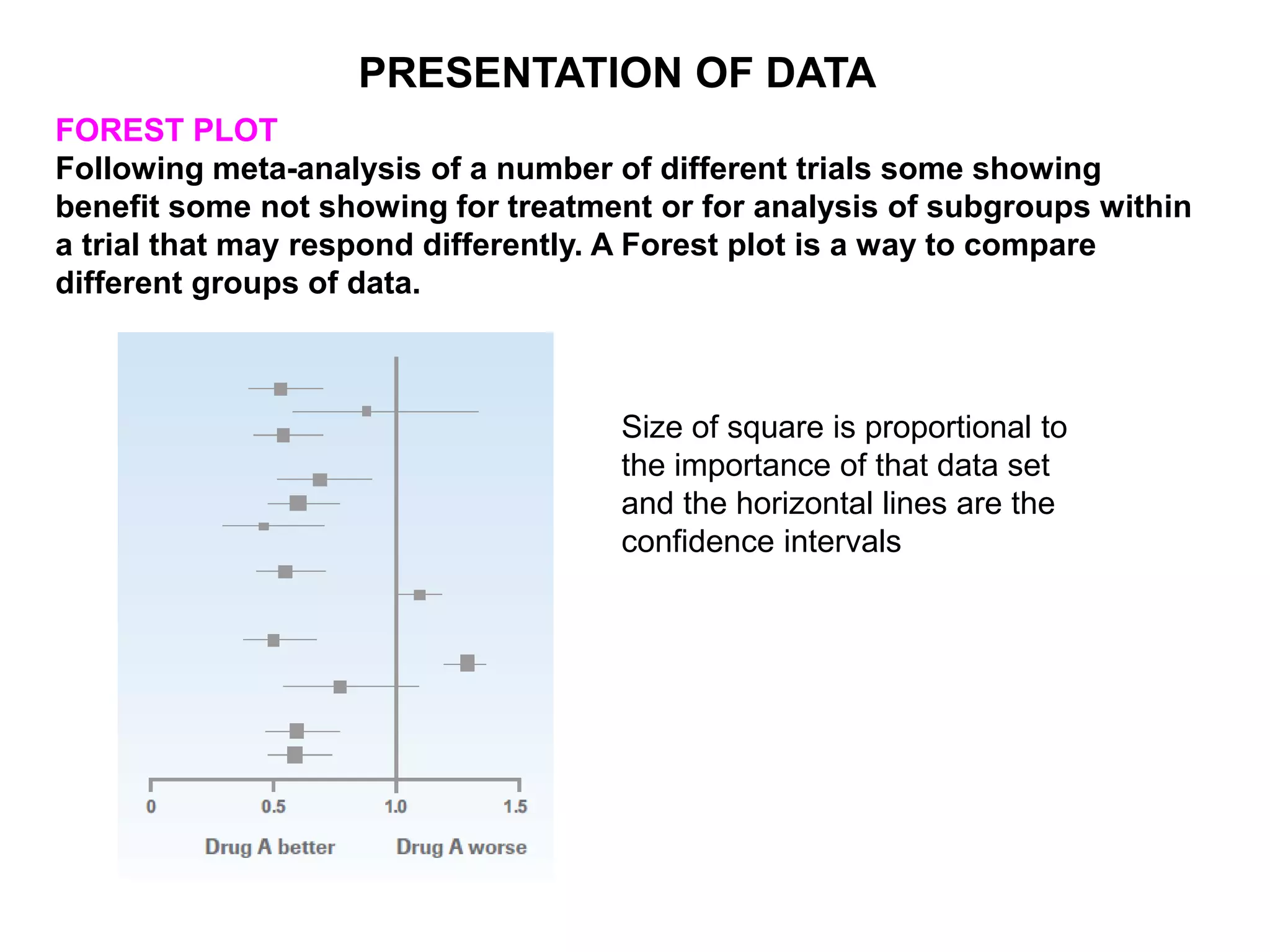
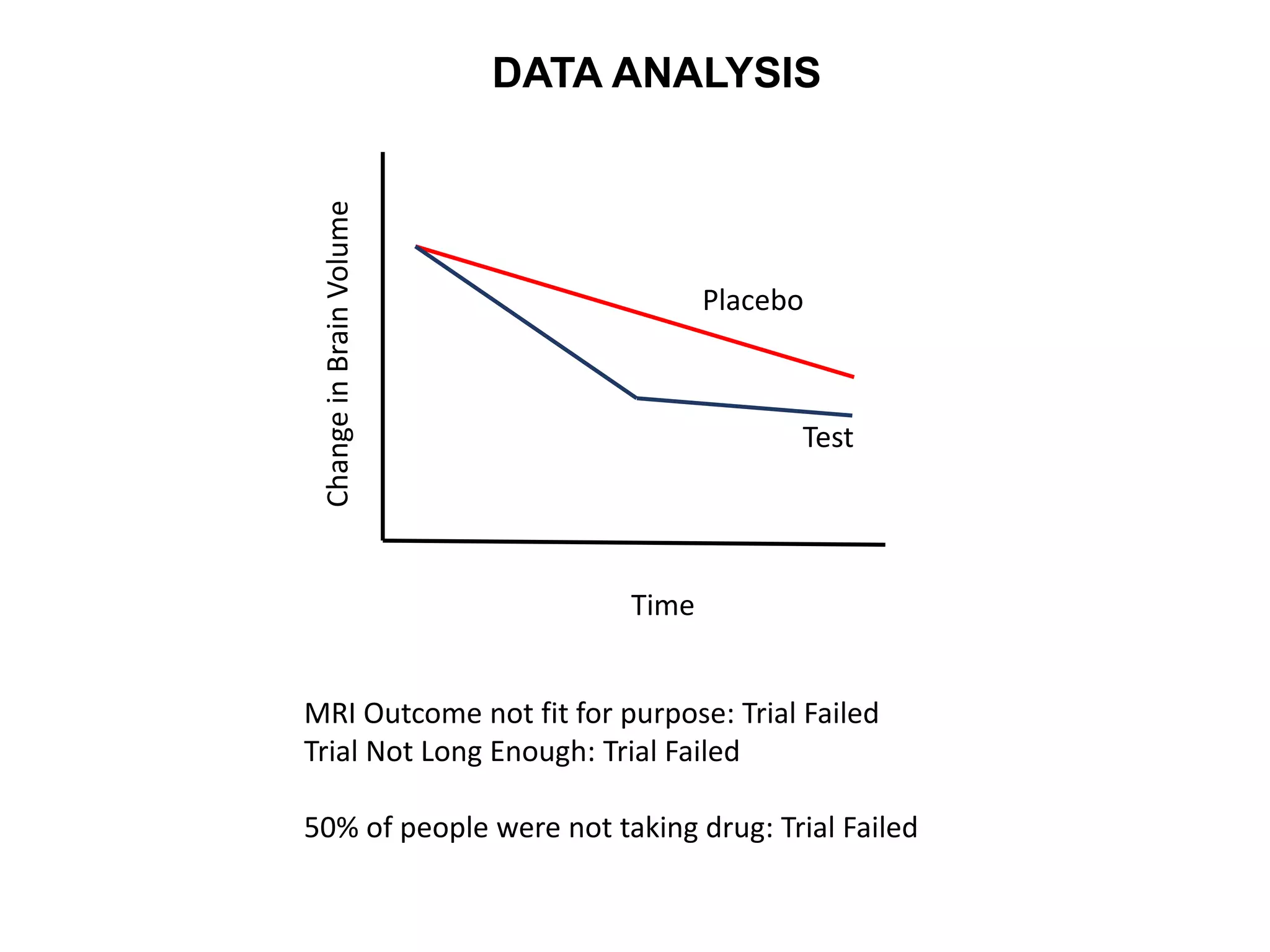
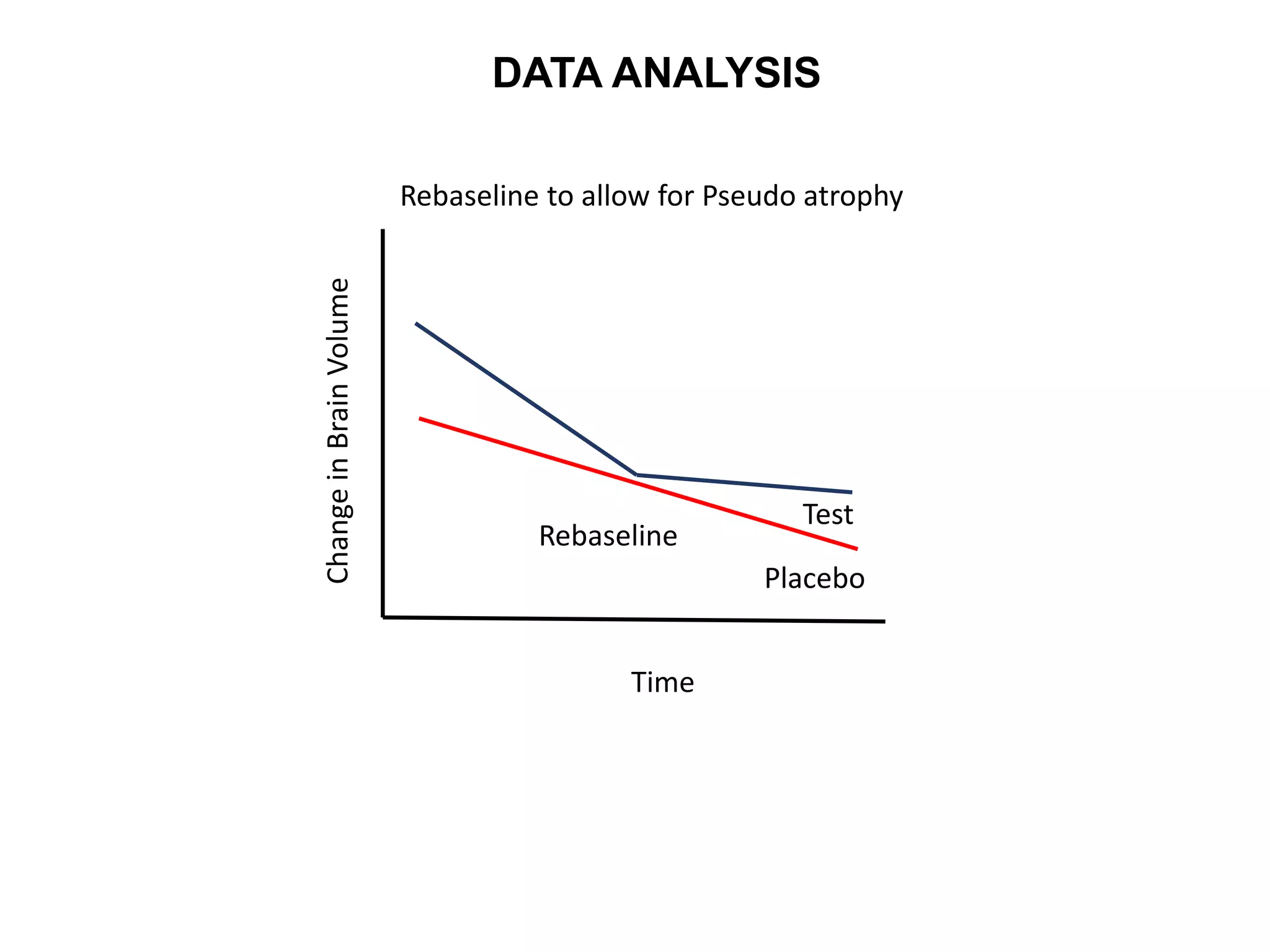
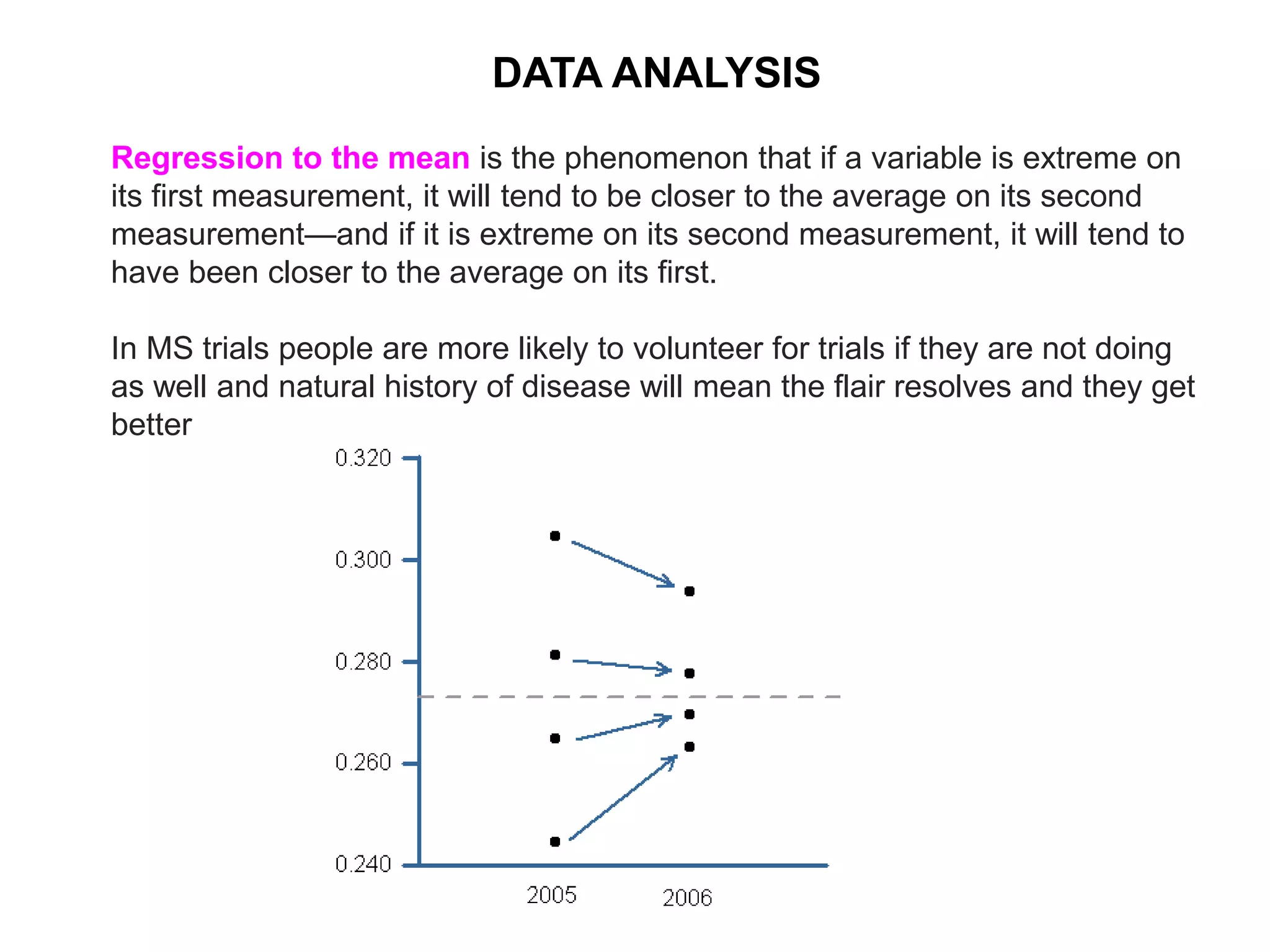
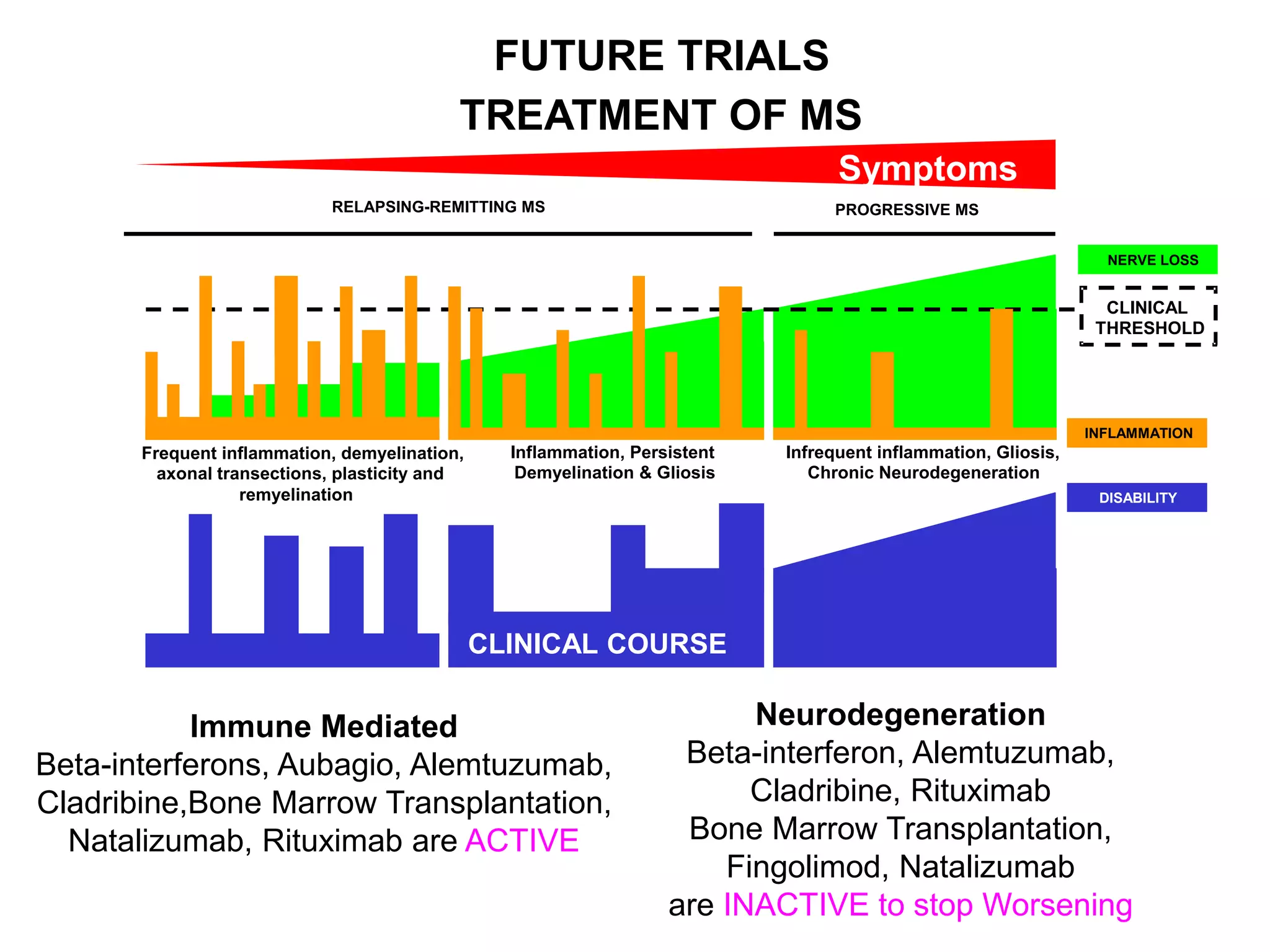
Clinical trials are scientific studies that test new drugs in human subjects. This document discusses the multi-phase clinical trial process, from pre-clinical animal studies through post-approval monitoring. It notes that trials progress from small Phase I safety studies in healthy volunteers to large Phase III efficacy trials in patients. The goal is to demonstrate a drug's benefits outweigh its risks before regulatory approval and marketing.