




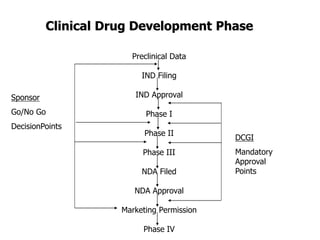
































![Phase IV Study / Clinical trial
• Helps to detect rare ADRs, Drug interactions
• Also to explore new uses for drugs [Sometimes called
Phase V]
• PERIODIC SAFETY UPDATE REPORTS
• To be submitted by the manufacturer every 6 months
for 2 yrs and then annually for next 2 yrs after
marketing approval](https://image.slidesharecdn.com/ctp-150215132923-conversion-gate02/85/Clinical-Trial-Phases-44-320.jpg)





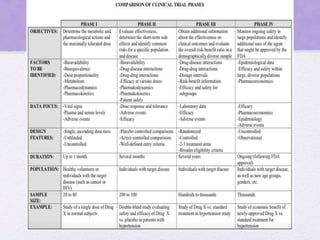





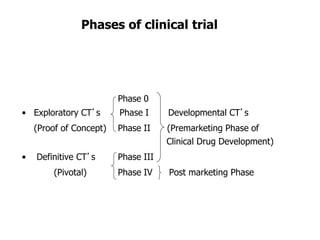
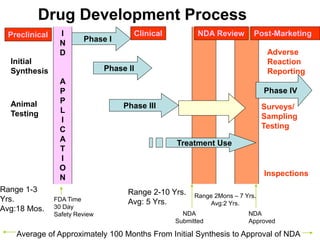
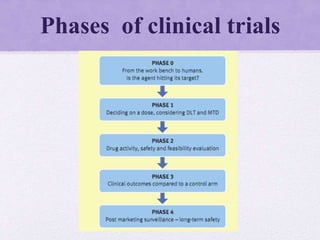
Clinical trials involve several phases: - Phase I trials involve small groups of healthy volunteers or patients and aim to determine the safety and tolerability of a new drug. - Phase II trials involve larger groups of patients and aim to determine efficacy and further evaluate safety. These trials provide preliminary data on effectiveness. - Phase III trials involve even more patients and aim to confirm effectiveness, monitor side effects, and compare the new treatment to standard treatment. These trials provide the primary data to support effectiveness. Regulatory approval is based on positive Phase III results showing safety and effectiveness.



























































































