Downloaded 698 times



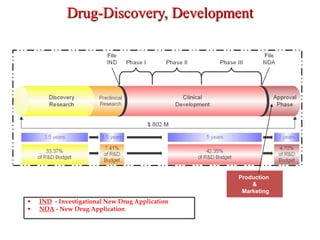
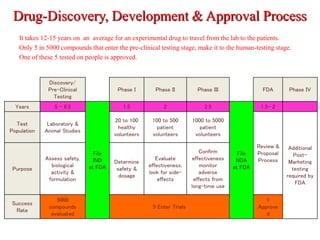






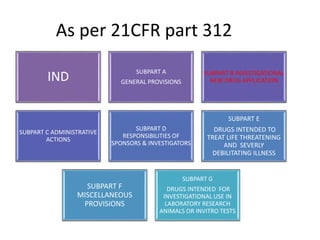



































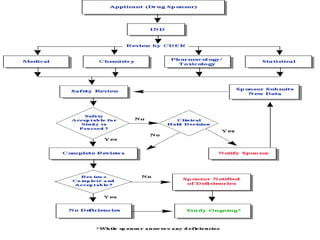

The document discusses an Investigational New Drug Application (IND), which is a submission to the FDA requesting permission to study an experimental drug in clinical trials. An IND provides safety data from animal studies and details of the proposed clinical trials. It includes information on the drug's chemistry, manufacturing, pharmacology and any previous human experience. The IND process allows a drug to move through preclinical and clinical testing in humans while being monitored by the FDA to ensure the protection of clinical trial subjects.
























![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)












![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)


























































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)










