


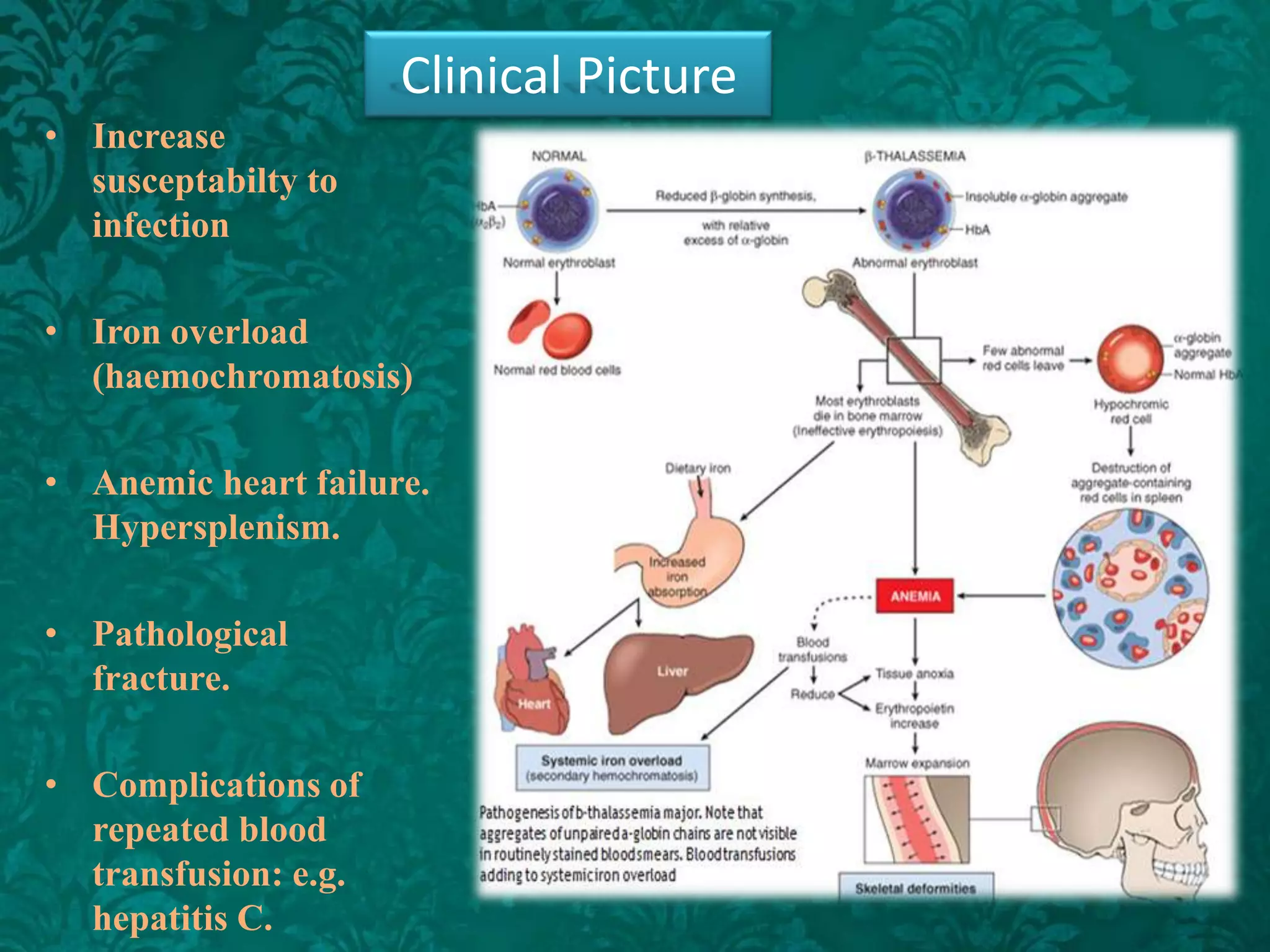
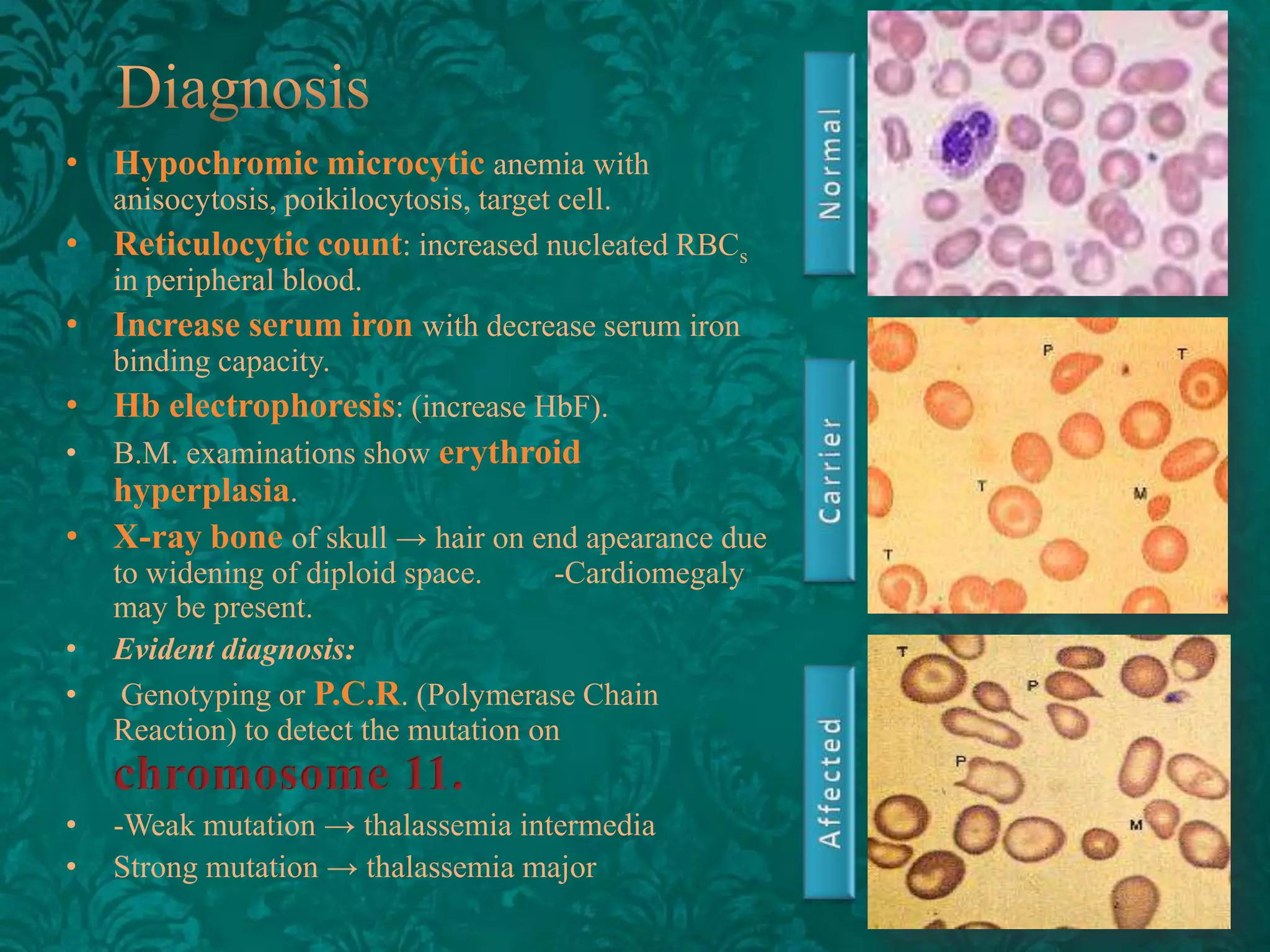
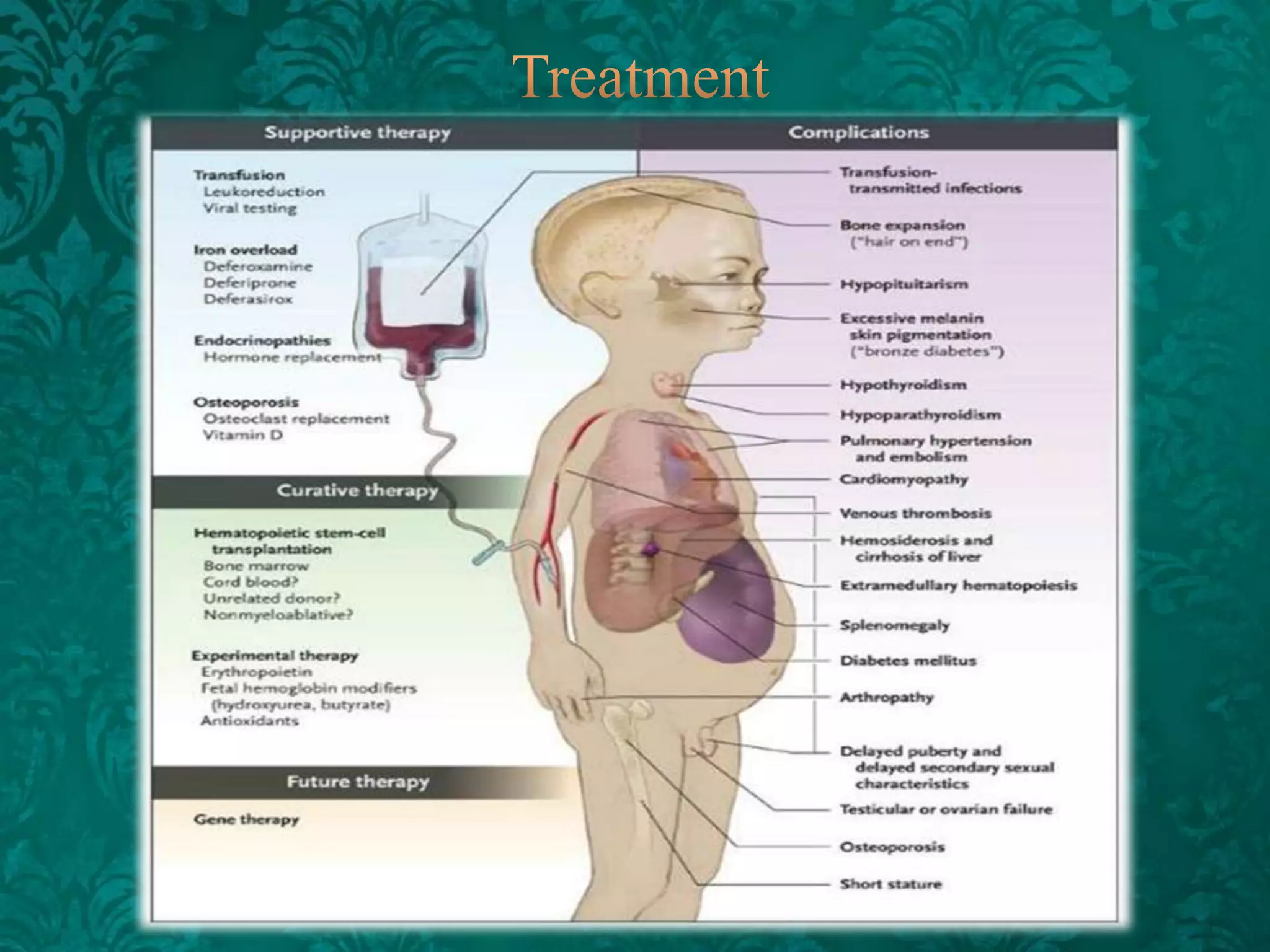




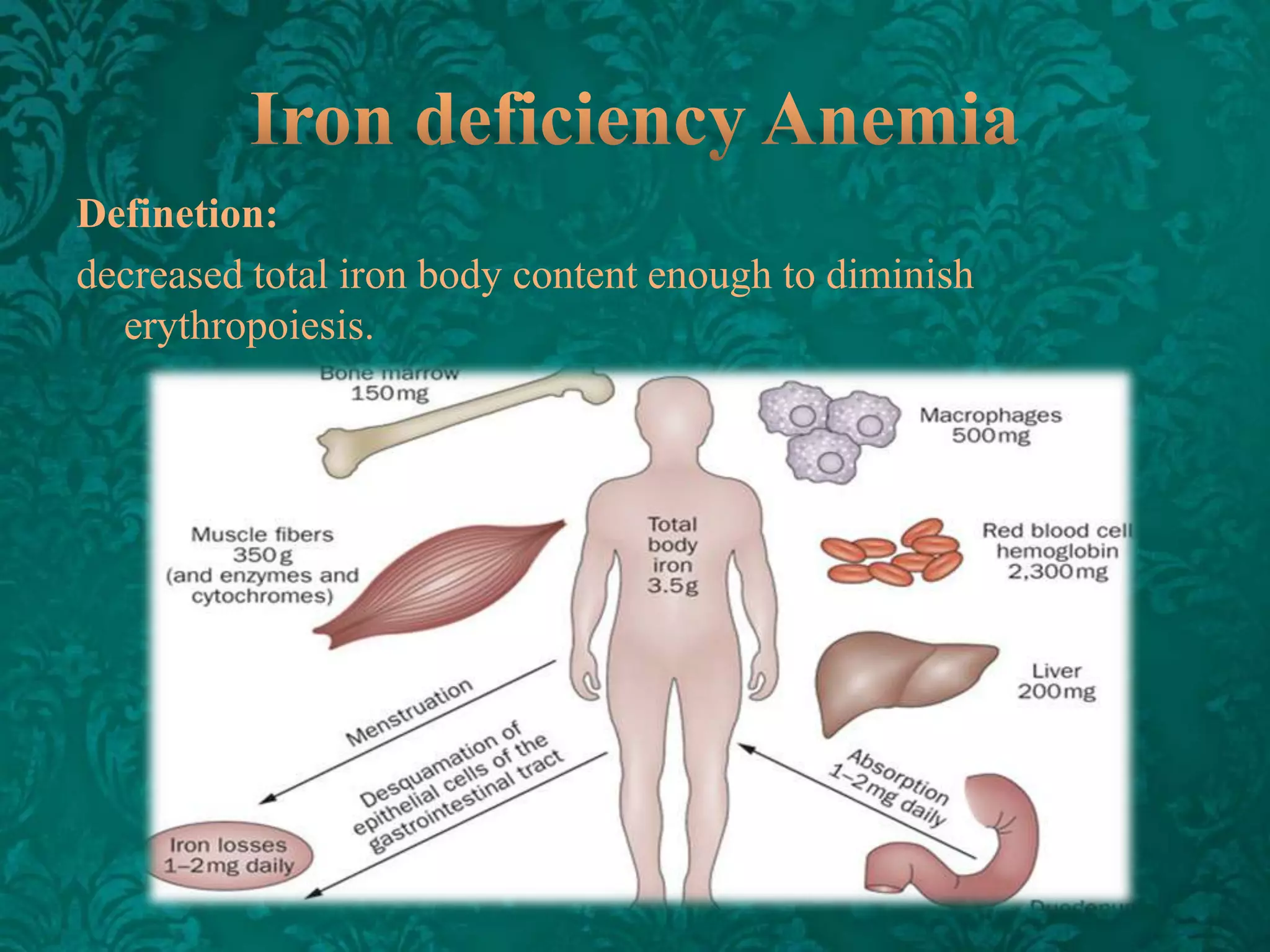

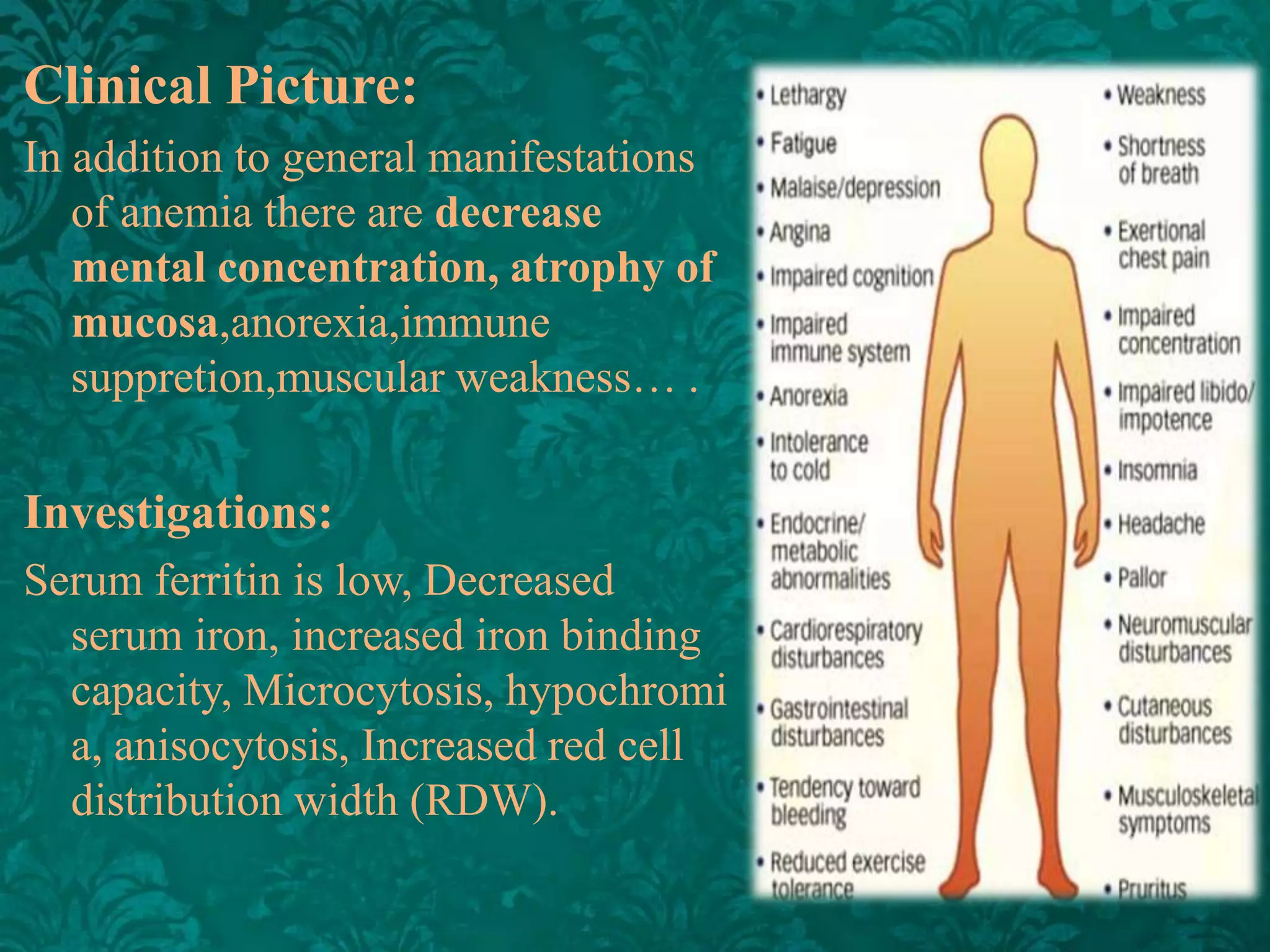







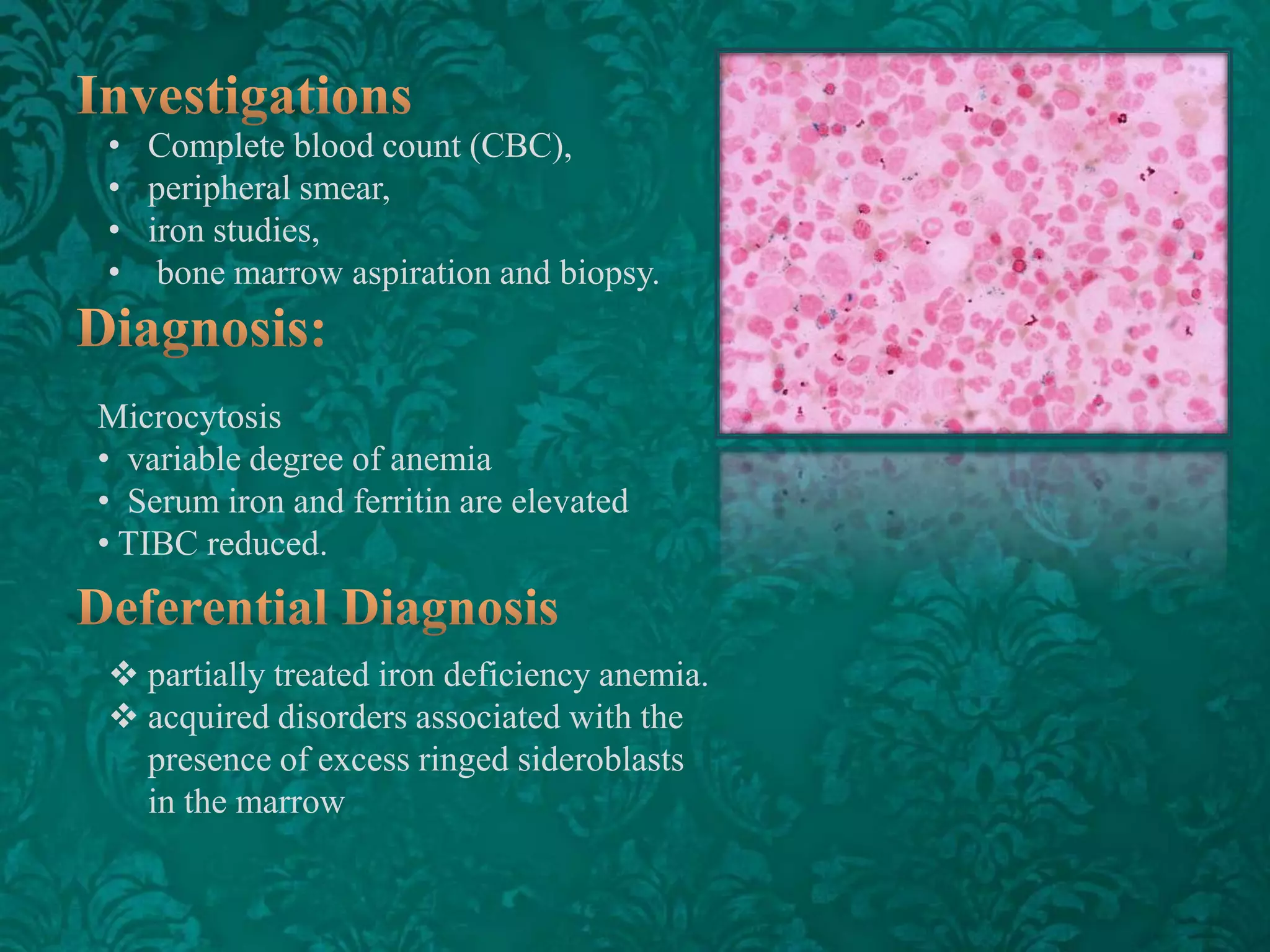




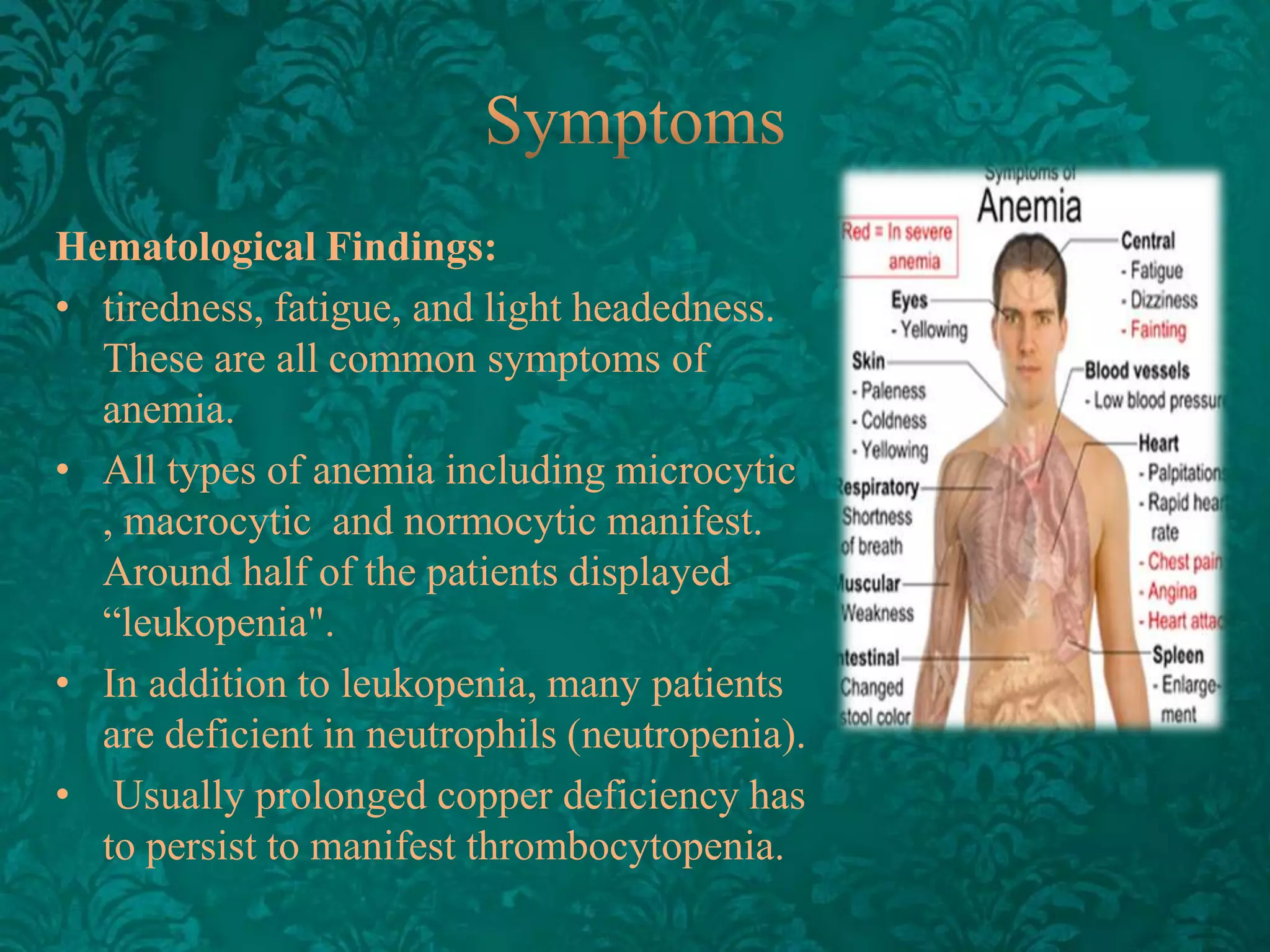




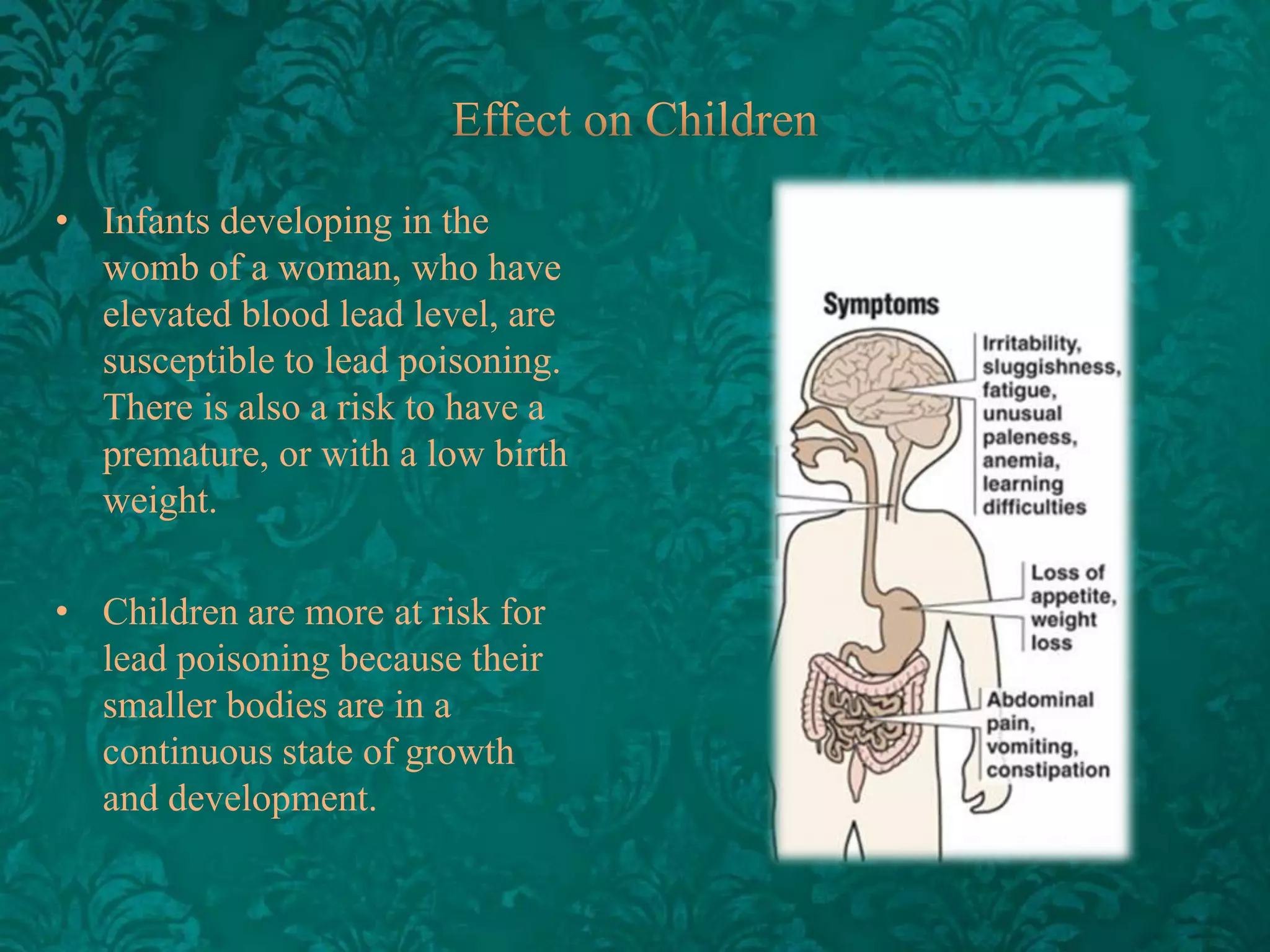

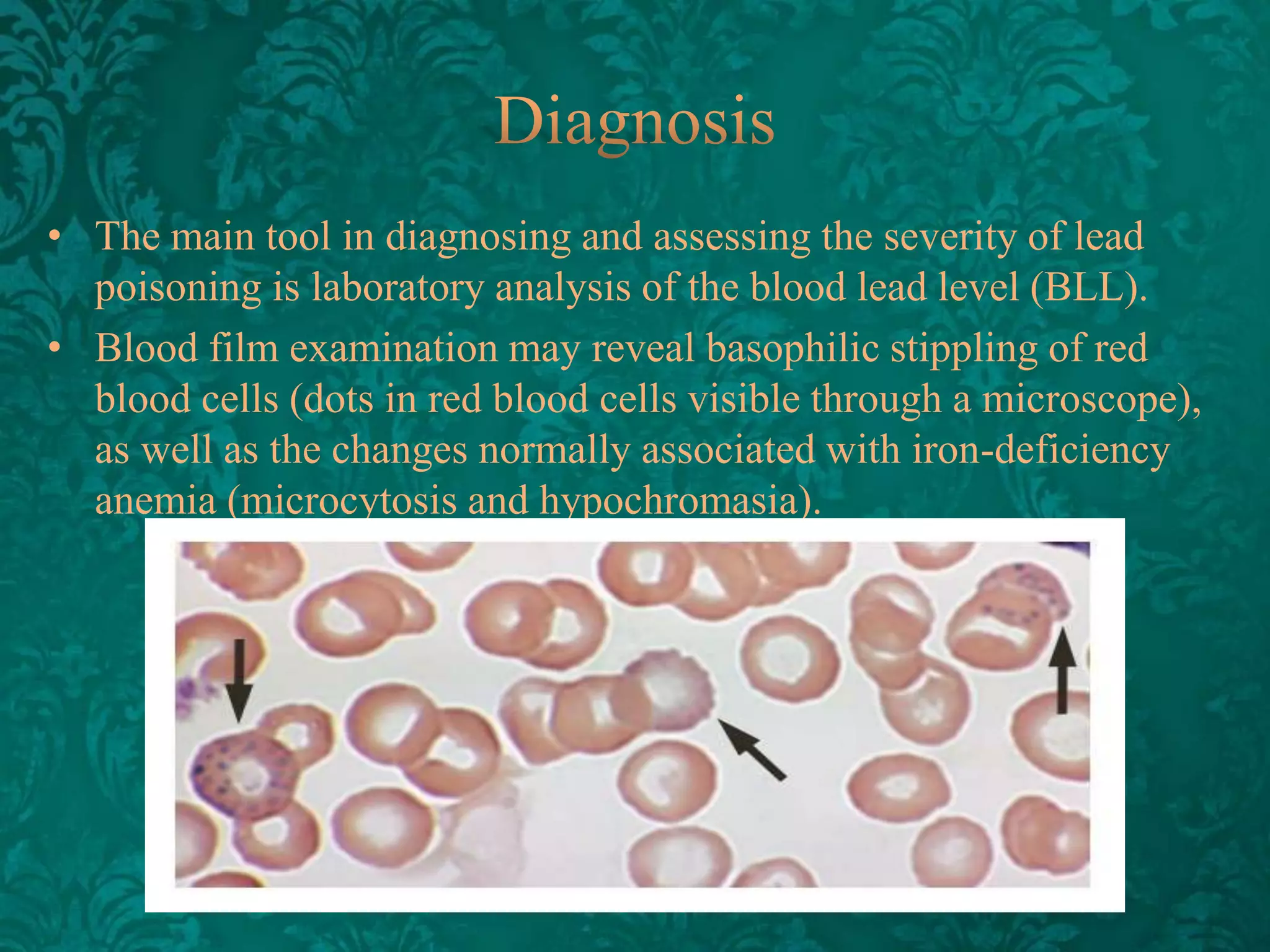

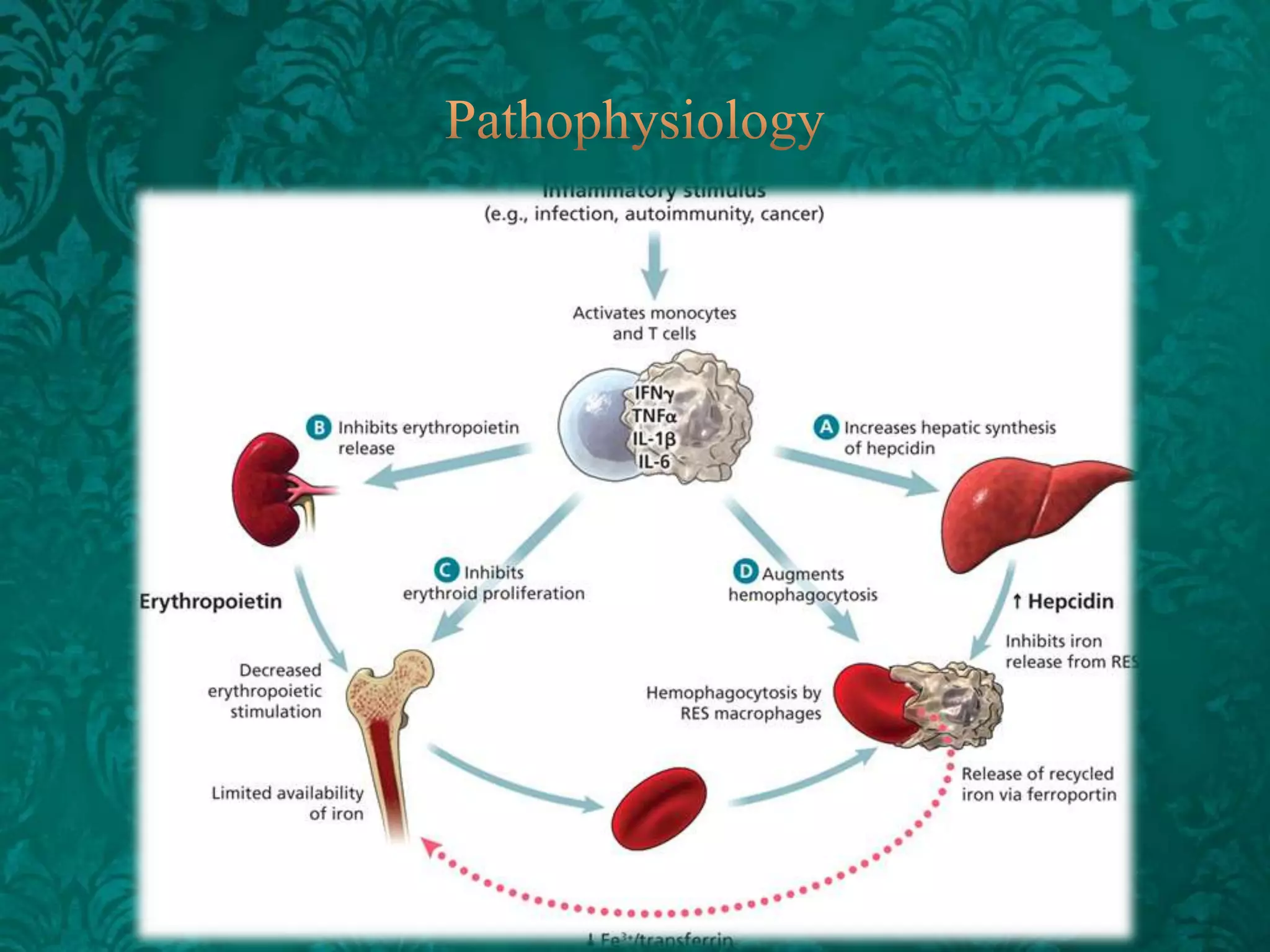



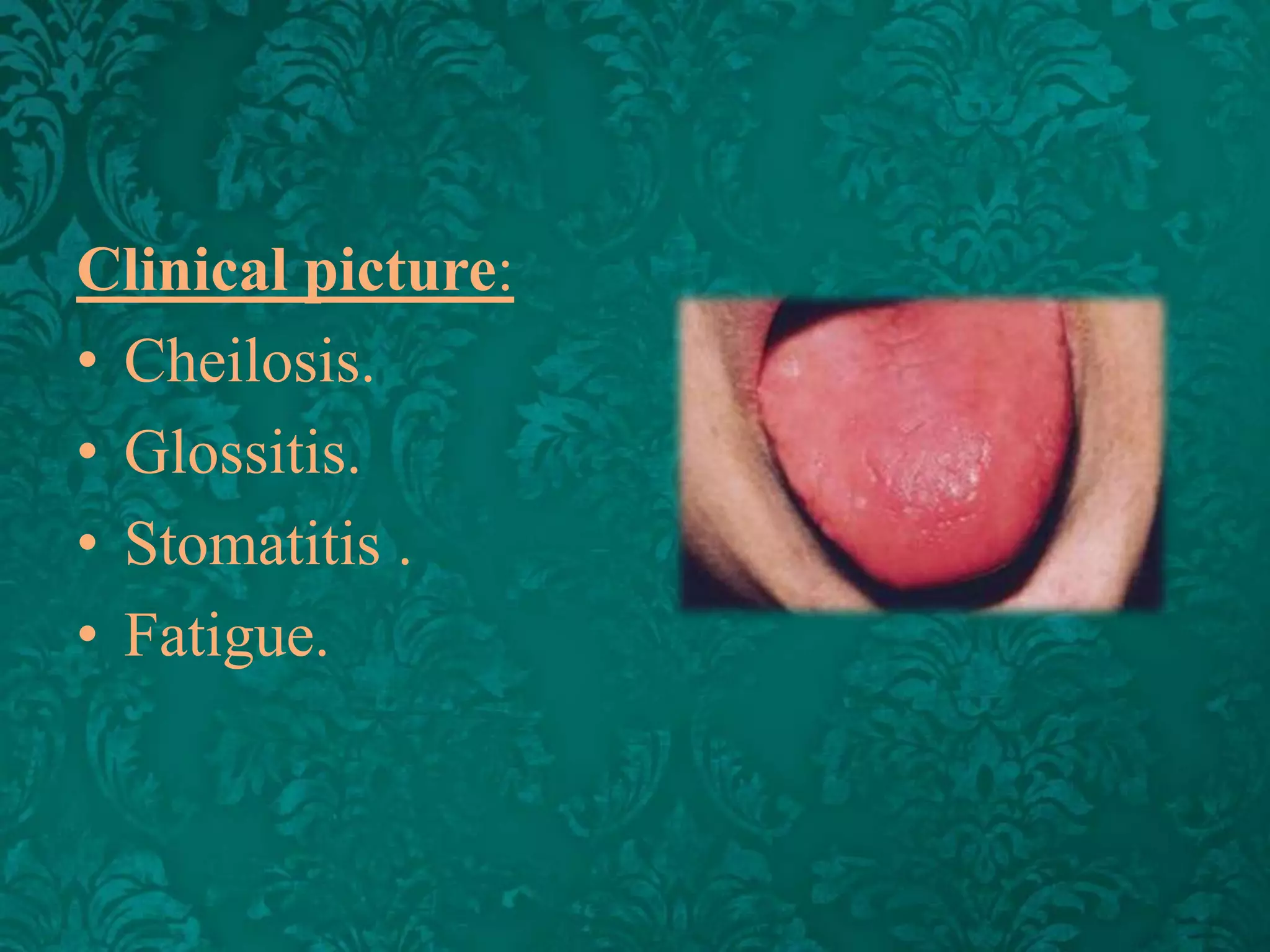



This document discusses β-thalassemia, a genetic blood disorder caused by mutations in the β-globin gene resulting in reduced or absent β-chain production and hemoglobin synthesis. It is characterized by microcytic hypochromic anemia and is most common around the Mediterranean sea. The degree of β-chain deficiency determines the severity from β° (no β-chains) to β++ (more β-chains). Clinical manifestations include anemia, jaundice, hepatosplenomegaly, skeletal abnormalities, and heart failure. Management involves blood transfusions, chelation therapy, and folic acid supplementation.


























































![Anaemia_ppt[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/anaemiappt1-230606161558-585643b1-thumbnail.jpg?width=640&height=640&fit=bounds)



































