Downloaded 76 times





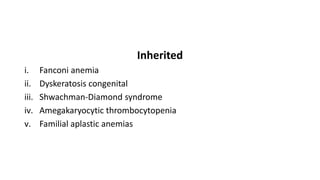













This document discusses aplastic anemia, a condition characterized by pancytopenia and bone marrow hypocellularity. It defines aplastic anemia, discusses its causes (acquired from things like radiation, drugs, viruses, immune diseases, or inherited genetic syndromes), epidemiology, pathophysiology, clinical features including symptoms and examination findings, diagnostic testing including blood tests and bone marrow biopsy, and treatment options including hematopoietic stem cell transplantation, immunosuppression, supportive care, and prognosis.



















































































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)


