Downloaded 131 times




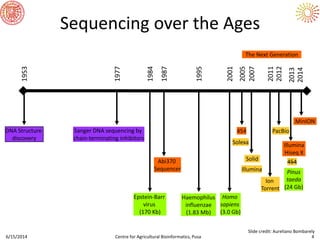
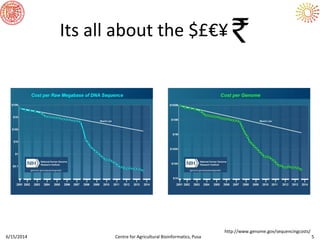


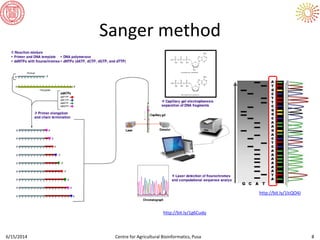





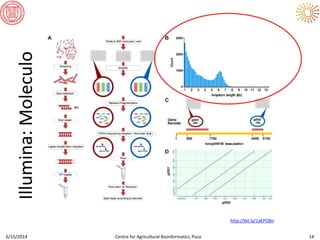

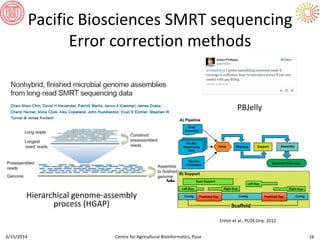
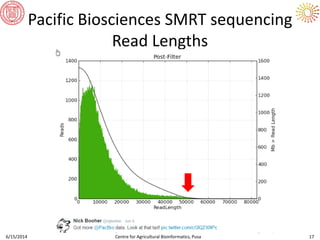



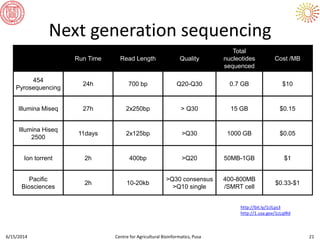
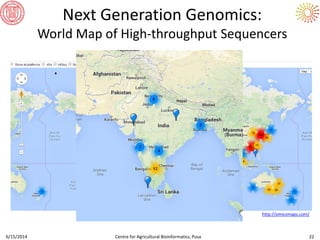
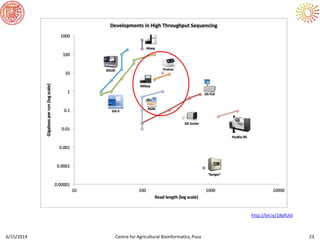
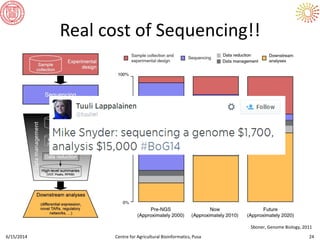

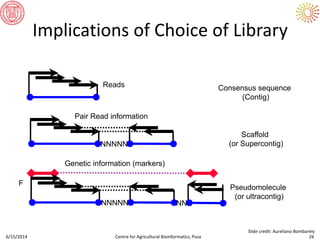
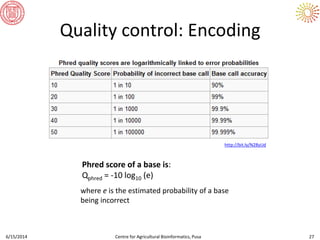

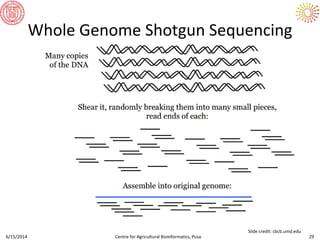
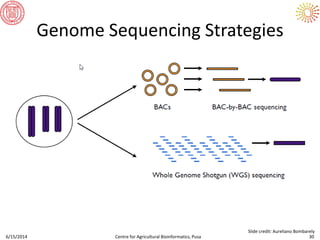
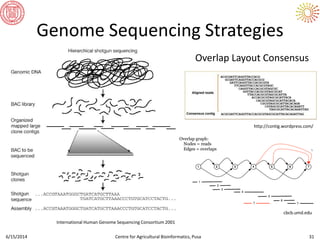
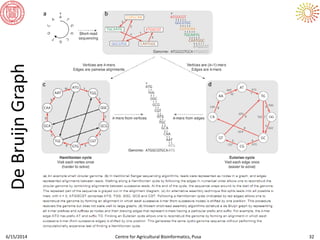
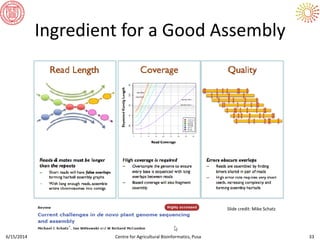
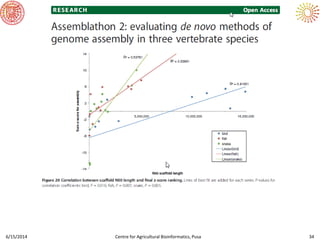
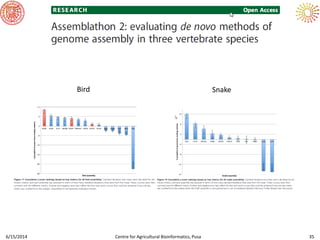

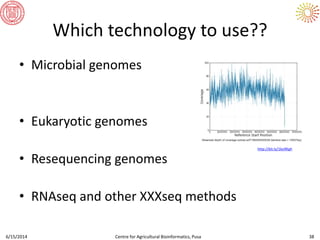

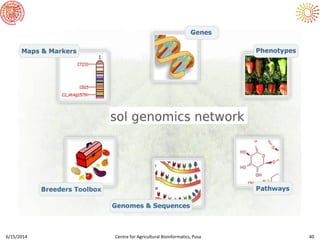

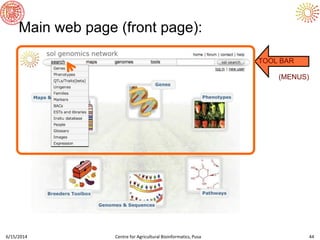
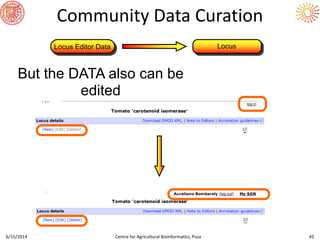
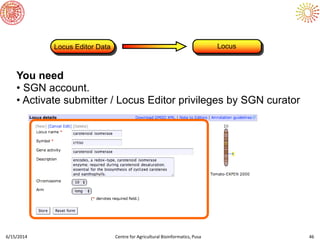
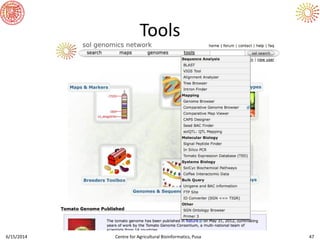
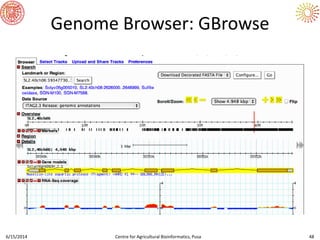



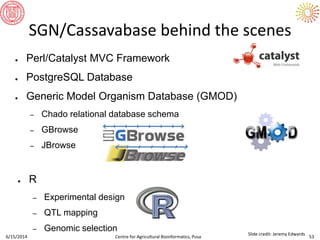

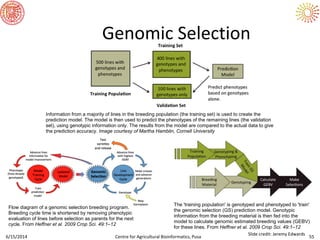

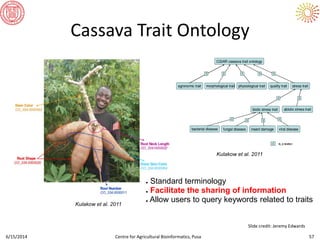



The document presents an overview of sequencing technologies and genome assembly processes, highlighting the evolution from Sanger sequencing to next-generation sequencing methods. It emphasizes the cost reductions and advancements in throughput over the years, detailing different sequencer types, their capabilities, and the implications for genomic research. Additionally, it discusses the integration of bioinformatics tools and data management systems in applications such as cassava breeding and genomic selection.


































































































